
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From energy to electron transfer photocatalysis (PenT → PET): oxidative cyclobutane cleavage alters the product composition†
Jens
Lefarth
 *a,
Alexander
Haseloer
b,
Lukas
Kletsch
b,
Axel
Klein
b,
Jörg
Neudörfl
a and
Axel G.
Griesbeck
*a
*a,
Alexander
Haseloer
b,
Lukas
Kletsch
b,
Axel
Klein
b,
Jörg
Neudörfl
a and
Axel G.
Griesbeck
*a
aUniversity of Cologne, Faculty of Mathematics and Natural Sciences, Department of Chemistry, Institute for Organic Chemistry, Greinstr. 4-6, Köln D-50939, Germany. E-mail: griesbeck@uni-koeln.de; Fax: +49-221-4701166
bUniversity of Cologne, Faculty of Mathematics and Natural Sciences, Department of Chemistry, Institute for Inorganic Chemistry, Greinstr. 4-6, Köln D-50939, Germany
First published on 28th October 2022
Abstract
Photocatalytic [2+2]-cycloadditions between cyclic enones and electron-rich cyclic enol ethers are initiated by triplet–triplet energy transfer from an excited iridium photocatalyst to the enone acceptor. The composition of the resulting cycloadduct mixture shows a surprising time dependency of the cyclobutane stereoisomeric ratio which indicates that the products are not photostable under the reaction conditions. The isomerisation of the cycloadducts 3 by a photoinduced redox process points to a switch from triplet energy transfer (PenT) to photoinduced electron transfer (PET) catalysis.
Photochemical [2+2]-cycloadditions can proceed by direct substrate activation through photon absorption via the first excited singlet or the energetically lower lying triplet state. The latter can also be generated by triplet–triplet energy transfer (PenT) using energetically adjusted sensitizers.1–5 Alternatively to these radical-type processes, photoinduced electron transfer (PET) is possible if appropriate electron donors or acceptors are present as substrates.6,7 Again, this PET route can be initiated by direct excitation of a substrate molecule or by use of a photoredox sensitiser. Obviously, in the photocatalysis of [2+2]-cycloadditions, PenT and PET can compete. The competition is controlled by energy arguments, i.e. appropriate triplet energies that allow exergonic PenT or excited state redox potentials that allow exergonic PET.
For practical reasons, photocatalytic syntheses are typically conducted until maximum conversion of the substrate that is used in lower amounts. The characteristic selectivity features such as regio- or stereoselectivity are consequently quite often determined only after complete or nearly complete conversions. These results are relevant for an overall reaction profile as well as for synthetic applications but might be misleading for a mechanistic description.
The problem that we address in this paper is the possible change in the mechanistic function of the photocatalyst that occurs after photoproduct formation using a properly designed photocatalyst (PC) for [2+2]-cycloaddition. As we have recently reported,8 the cycloaddition between chalcone (1) and 2,3-dihydrofuran (2,3-DHF) (2) photocatalysed by [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (abbreviated as [Ir-F], Scheme 1) results in four cyclobutane diastereomers with the compound 3a as the major product after moderate conversions (Scheme 1). Likewise, cyclic enones and electron-rich alkenes give cycloadducts in high yields and good stereoselectivities. Redox processes that involve the enones as acceptors or the alkenes as donors are energetically improbable. On the other hand, triplet state energy comparison resulted in an exergonic energy transfer mechanism as the most likely activation meachanism. Herein, we would like to report on the unexpected isomerisation of diastereomers that was observed after prolonged irradiation of the reaction mixture.
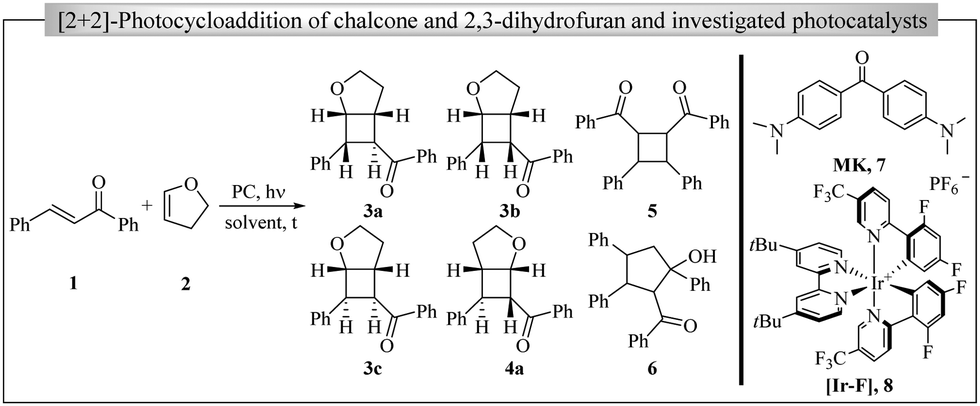 | ||
| Scheme 1 Model reaction for the photosensitised [2+2]-cycloaddition of acceptor–donor systems and investigated photocatalysts. | ||
During this study, we focussed on two potential photocatalysts (Table 1): Michler's ketone (7, MK)9–11 and [Ir-F] (8).12,13 In the first part, the hypothesis of a time-dependent secondary PET reaction is proposed based on the experimental observations on the product selectivities of cyclobutanes 3a and 3b as well as dimers 5 and 6 (Scheme 1). In the second part, this theory will be underlined by cyclic voltammetry (of 3a and 3c) and crystal structure analysis (3c) as well as additional cyclobutane products (Fig. 1).14
 | ||
| Fig. 1 Cyclobutanes analysed by cyclic voltammetry and crystal structure analysis (except 3a and 12). | ||
During our previous study,8 we observed that cyclopentanol dimer 6, which was postulated to be formed by a P(C)ET mechanism,15 seemed to be the preferred product from MK (Table 2, entry 2), whereas the cyclopentanol 6 was only a minor by-product with [Ir-F] as the photocatalyst. Based on the redox potentials shown in Table 1, this correlates to an exergonic oxidative quenching mechanism of MK by chalcone (Ered = −1.48 V vs. SCE; ET = 2.17 eV),16 whereas oxidative quenching for [Ir-F] was endergonic. However, further optimisation studies for the reaction with [Ir-F] as the photocatalyst revealed different product ratios after an irradiation period of 4 h (entry 5) compared to a much longer period of 24 h (entry 4). When using MK, however, a comparison of the reaction times was difficult to obtain due to a very low conversion of only 9% that was like the non-sensitised reaction after 4 h. In case of the non-sensitised reaction, no significant deviation in the product ratios was observed when irradiated for 4 h, 24 h or 48 h regardless of the excitation wavelength (350 or 455 nm). Therefore, we concluded that the different product ratios correlate to a secondary reaction from the cyclobutane products by PET similar to a photocycloreversion reaction.17,18 Additionally, the photolysis experiments were performed in different solvents, which will only be illustrated for the case of MeCN and toluene (see Tables S1–S3, ESI,† for additional information). From non-polar solvents such as toluene, a general observation was a significantly preferred formation of the chalcone dimers, which may be attributed to an increased π-stacking interaction of the substrates and solvent molecules. In addition, an increased formation of cyclobutane 3b was observed in case of the non-sensitised reaction (entry 6) as well as for the reaction with MK (entry 7) compared to the reaction in MeCN. The experiments with [Ir-F] as the photocatalyst, however, again showed different product ratio between cyclobutane 3a and 3b with varying reaction time. For these experiments, similar product ratios were obtained when compared with the results for MeCN after 4 h (entry 5 and 9) and after 24 h (entry 4 and 8).
| No. | PCa | Solvent | Yield [%]b | Product ratioc | ||||
|---|---|---|---|---|---|---|---|---|
| 3a | 3b | 5 | 6 | 3a/3b | ||||
| a Conditions: if not stated otherwise, irradiation for 24 h at 350 nm (for uncatalyzed and Michler's ketone [MK] reactions) or 455 nm (for [Ir-F] (1 mol%) reactions). b GC-MS analysis for conversion to cyclobutanes 3 and 4 and dimers 5 and 6. c Ratios were referenced to the major isomer 3a (set to 1.0). d Mean value of four experiments. e Irradiated for 4 h. | ||||||||
| 1d | — | MeCN | 51 | 1.0 | 0.34 | 0.11 | — | 2.94 |
| 2 | MK | MeCN | 97 | 1.0 | 0.50 | 0.02 | 1.58 | 2 |
| 3e | MK | MeCN | 9 | 1.0 | 0.21 | 0.24 | 0.79 | 4.76 |
| 4 | [Ir-F] | MeCN | >99 | 1.0 | 1.35 | 0.01 | 0.03 | 0.74 |
| 5de | [Ir-F] | MeCN | 98 | 1.0 | 0.28 | 0.20 | 0.08 | 3.57 |
| 6 | — | Toluene | 83 | 1.0 | 0.89 | 1.63 | — | 1.12 |
| 7 | MK | Toluene | 95 | 1.0 | 15.3 | — | 70.8 | 0.07 |
| 8 | [Ir-F] | Toluene | 99 | 1.0 | 1.62 | 0.55 | 0.57 | 0.62 |
| 9e | [Ir-F] | Toluene | 73 | 1.0 | 0.29 | 1.79 | 0.25 | 3.45 |
Even though the experimental data already indicated that the observations are likely correlated to a PET process, a possible thermal instability of the photoproducts could also attribute to our findings. The reported results, however, were reproduced several times (up to ten times for some experiments), indicating that a spontaneous radical-induced reaction is improbable. In addition, the cyclobutane products in the dark showed high stability over several months with only minor degradation. Hence, a secondary photochemical reaction from the cyclobutane products appeared to be the most viable explanation for our observations.
In the next step, we wanted to study the energy profile of such a secondary PET process and determined redox potentials for 3a and 3c in comparison with the reported products (Fig. 1 and 2 and Fig. S1–S7, ESI†) using cyclic voltammetry.8,19
 | ||
| Fig. 2 Cyclic voltammograms of cyclobutane 3a in nBu4NPF6/MeCN. | ||
All oxidation and reduction processes were irreversible due to rapid follow-up reactions (see our mechanistic considerations below). Since the peak potentials of irreversible peaks are depending on the solvent, scan rate and concentration, we chose the same conditions for all samples and determined offset potentials as shown in Fig. 2, which approximate the half-wave potentials and give a good estimate of E0. The CV of the cyclobutane 3a (Fig. 2) showed an unexpectedly low oxidation potential Eoffset(ox) of 1.2 V vs. ferrocene/ferrocenium (Table 3) alongside with 9c and 13. Product 3c lies slightly higher with 1.43 V, which is in the range of cyclobutanes 9a, 10, 11, and 12. Taken together with the reduction potentials, the electrochemical gap of 3a and 3c is about 3.6 eV. Again, the cyclobutane 13 shows a similar value, while 9a, 10, 11, and 12 show gap energies of more than 4.5 eV. As can be expected from the electron-donating MeO substituent in 9c and the electron-demanding CF3 group in 9b, the oxidation potential increases massively along the the series 9c < 9a < 9b.
| E pa(ox) | E offset(ox) | E pc(red) | E offset(red) | ΔE | |
|---|---|---|---|---|---|
| a Measured in 0.1 M nBu4NPF6/MeCN at 298 K, 100 mV s−1 scan rate at a conc. of 7 mM. Anodic peak potential Epa for irreversible oxidations, cathodic peak potentials Epc for irreversible reductions, measured with an accuracy of ±0.005 V and offset potentials Eoffset (as defined in Fig. 2) in V vs. ferrocene/ferrocenium. ΔE = electrochemical gap between HOMO and LUMO. b No defined peaks observed, maximum Eoffset potentials estimated (see ESI). | |||||
| 3a | 1.29 | 1.20 | −2.45 | −2.38 | 3.58 |
| 3c | 1.57 | 1.43 | −2.43 | −2.34 | 3.77 |
| 9a | 1.63 | 1.46 | —b | −3.15 | 4.58 |
| 9b | —b | >1.55b | —b | <−2.97b | 4.52 |
| 9c | 1.08 | 1.03 | —b | <−3.12b | 4.15 |
| 10 | 1.75 | 1.65 | —b | <−3.29b | 4.94 |
| 11 | 1.83 | 1.56 | —b | <−3.24b | 4.80 |
| 12 | —b | 1.53 | —b | <−3.17b | 4.70 |
| 13 | 1.19 | 1.10 | −2.31 | −2.24 | 3.43 |
Although an estimation of the Gibbs energy20 for 3a for a PET appeared to be slightly endergonic with [Ir-F] (1[Ir-F]: ΔGPET ∼7 kcal mol−1 using the redox potentials for the singlet excited Ir-F and 3a, normalized to the standard calomel electrode and Ecoulombic = 0), a PET reaction is still possible. For instance, substrate activation by components of the reaction mixture or slightly increased temperatures from photolysis (∼30 °C) could lower the activation energy. Moreover, a significant difference between the oxidation potentials of cyclobutanes 3a and 3c (∼0.3 V) was observed that might explain the sole conversion of cyclobutane 3a after prolonged irradiation in the presence of [Ir-F]. Spectroelectrochemical measurements on the oxidation reaction were performed for cyclobutane 3a and 3c (Fig. 3).
 | ||
| Fig. 3 UV-vis absorption spectra during electrochemical oxidation of cyclobutane 3a (left) and 3c (right) in nBu4NPF6/MeCN. | ||
In both cases, a new absorption maximum appeared on electro-chemical oxidation at λmax = 400 nm and around λmax = 305 nm that indicate the formation of a product with an extended π-system. Unfortunately, both processes turned out to be irreversible, since on return-scan we did not observe the starting materials. In the next step, we wanted to gain insight into if the oxidation event is followed by C–C bond cleavage. For this, we compared the C–C bond lengths of the studied cyclobutane 3c from a crystal structure determination with those of further cyclobutane products (Fig. 1).
We hypothesised that the oxidation weakened the σ-bond of the cyclobutane moiety (bond 1 or bond 4, numbering see Fig. 1), resulting in the more stable radical cation intermediate. For comparison, the unsubstituted cyclobutane has a bond length of 1.556(1) Å,21 whereas the octamethylcyclobutane has a bond length of 1.571 Å (by AM1).22 In contrast, exceptionally long bond lengths (1.61–1.72 Å) have also been observed for e.g. phenyl substituted cyclobutanes and anthracene dimers.23,24 From our analysis, however, such drastic elongations were not observed (Table 4). Nevertheless, in almost every case, one bond showed a significant elongation compared to the other bonds of the cyclobutane moiety. Cyclobutane 3c shows a slight elongation of bond 1 (1.584(2) Å) compared to the other bonds (∼1.55 Å). For cyclobutane 13, bond 4 is elongated (1.579(2) Å). However, even though cyclobutane 11 showed a markedly higher oxidation potential, the same elongation of bond 4 was observed (1.585(1) Å). In this case, we attribute this to steric repulsion between the methyl and phenyl substituents at C1 and C4. Nevertheless, it may be possible, for cyclobutanes that relate to our investigated molecular structure, to correlate bond elongations for cyclobutanes with lower oxidation potentials to electronic rather than steric effects.22 In fact, this can also be seen from the comparison of three cyclohexenone-based cyclobutanes (9a–c). All three substrates have a similar molecular structure, but a slightly altered electronic structure from 4-phenyl substitution. Therein, the most electron-rich cyclobutane 9c shows the largest bond elongation compared to the least electron-rich cyclobutane 9b.
| Bond | 3c | 13 | 11 | 9a | 9b | 9c_1 | 9c_2 | 9c_3 | 9c_4 |
|---|---|---|---|---|---|---|---|---|---|
| a From single crystal X-ray diffraction analysis. For numbering, see Fig. 1). Values were rounded with a margin of error of ±1–2 on the last digit (±4 for 9c), precise data in Table S5 (ESI). b Four crystallographic independent structures were obtained per asymmetric unit. | |||||||||
| 1 | 1.584 | 1.555 | 1.555 | 1.566 | 1.558 | 1.574 | 1.568 | 1.571 | 1.574 |
| 2 | 1.547 | 1.563 | 1.567 | 1.567 | 1.569 | 1.566 | 1.567 | 1.564 | 1.564 |
| 3 | 1.555 | 1.551 | 1.562 | 1.549 | 1.554 | 1.547 | 1.551 | 1.551 | 1.549 |
| 4 | 1.558 | 1.579 | 1.585 | 1.563 | 1.564 | 1.571 | 1.577 | 1.577 | 1.575 |
Most noticeably, however, was the correlation of the dihedral angles and the redox potentials (Table 5). The lower the oxidation potential was, the more planar the cyclobutane-core seemed to be. This can further be correlated to the electron density of the products. For cyclobutane 13 (highest electron density), the lowest oxidation potential and a small dihedral angle was observed (4.9°). In contrast, cyclobutane 11 (least electron-dense cyclobutane), has the largest dihedral angle and oxidation potential. This is confirmed by the series of cyclohexenone–cyclobutanes 9a–c. A methoxy substitution resulted in reduced dihedral angles (∼5.2°), whereas CF3-substitution led to larger dihedral angles (12.8°). Further data, such as additional bond lengths and bond angles can be found in Tables S4–S6, ESI.†
| Compound | 3c | 9a | 9b | 9c_1 | 9c_2 | 9c_3 | 9c_4 | 13 | 11 |
|---|---|---|---|---|---|---|---|---|---|
| a From single crystal X-ray diffraction analysis. Mean values from the four individual cyclobutane angles are given. Precise data can be found in Table S6 (ESI). b From cyclic voltammetry (see Table 3). | |||||||||
| Dihedral angle [°] | 5.4 | 5.8 | 12.8 | 5.7 | 5.3 | 4.5 | 5.3 | 4.9 | 22.2 |
| E offset(ox) [V] | 1.43 | 1.46 | >1.55 | 1.03 | 1.03 | 1.03 | 1.03 | 1.10 | 1.56 |
Based on the data obtained from the reaction of chalcone (1) and 2,3-dihydrofuran (2), we postulate a mechanism that involves the oxidation of bond 1 by a reductive quenching of [Ir-F]* (Scheme 2). The so formed radical cation is anticipated to undergo σ-bond rotation to form the thermodynamically most stable conformation. After reversed electron-transfer (RET) by the [Ir-F]˙− radical anion, the cyclobutane is regenerated. For this process to occur, rotation processes as well as the RET has to occur in a short time-interval so that contact ion pair separation cannot take place. In contrast, oxidation of bond 4 would, after RET, lead to a new diastereomer 3d with a loss in stereoinformation at C1 and C4. Since the formation of such a stereoisomer was not observed, the oxidation of bond 1 seems to be most likely.
 | ||
| Scheme 2 Postulated mechanistic Scheme for a secondary PET reaction of cyclobutane photoproducts from the [Ir-F] photocatalyst. | ||
In summary, we reported on the secondary photoreaction of the [2+2]-cycloaddition photoproducts of π2(Acc)–π2(Do) systems that we attributed to a PET process from the iridium photocatalyst. Whereas such processes have previously been observed in e.g. cycloreversion processes,18 we were only able to observe a diastereoisomerization process instead of cycloreversion. This observation was only made after prolonged irradiation of the reaction mixture and highlights the complexity of photocatalytic reactions. While screening until complete conversion may be a practical method, it assumes that the photoproducts are stable. However, under highly energetic photocatalytic conditions, such assumptions must be verified by additional experiments under prolonged irradiation. Although we did so far not implement our findings in possible synthetic transformations for direct derivatisation of these cyclobutane products, such a secondary oxidation event could likely be utilised to insert nucleophiles directly into the photoproducts.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Bach, Synthesis, 1998, 683–703 CrossRef CAS.
- S. Poplata, A. Troster, Y. Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
- M. J. James, J. L. Schwarz, F. Strieth-Kalthoff, B. Wibbeling and F. Glorius, J. Am. Chem. Soc., 2018, 140, 8624–8628 CrossRef CAS PubMed.
- D. Sarkar, N. Bera and S. Ghosh, Eur. J. Org. Chem., 2020, 1310–1326 CrossRef CAS.
- Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS PubMed.
- N. E. S. Tay, D. Lehnherr and T. Rovis, Chem. Rev., 2022, 122, 2487–2649 CrossRef CAS PubMed.
- M. Neumann and K. Zeitler, Chemistry, 2013, 19, 6950–6955 CrossRef CAS PubMed.
- J. Lefarth and A. G. Griesbeck, J. Org. Chem., 2022, 87, 8028–8033 CrossRef CAS PubMed.
- H. J. Timpe, K. P. Kronfeld, U. Lammel, J. P. Fouassier and D. J. Lougnot, J. Photochem. Photobiol., A, 1990, 52, 111–122 CrossRef CAS.
- J. Luo and J. Zhang, J. Org. Chem., 2016, 81, 9131–9137 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- N. A. Till, L. Tian, Z. Dong, G. D. Scholes and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 15830–15841 CrossRef CAS PubMed.
- Since compound 3b could only be isolated as a liquid and as a mixture with compound 3a, further studies were performed with compound 3c as a reference system instead of 3b.
- B. Kurpil, Y. Markushyna and A. Savateev, ACS Catal., 2019, 9, 1531–1538 CrossRef CAS.
- T. Lei, C. Zhou, M. Y. Huang, L. M. Zhao, B. Yang, C. Ye, H. Xiao, Q. Y. Meng, V. Ramamurthy, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 15407–15410 CrossRef CAS PubMed.
- K. Okada, K. Hisamitsu and T. Mukai, Tetrahedron Lett., 1981, 22, 1251–1254 CrossRef CAS.
- T. Hartman, M. Reisnerova, J. Chudoba, E. Svobodova, N. Archipowa, R. J. Kutta and R. Cibulka, ChemPlusChem, 2021, 86, 373–386 CrossRef CAS PubMed.
- J. Lefarth, J. Neudörfl and A. G. Griesbeck, Molbank, 2021, 2021, M1256 CrossRef.
- A. D. McNaught and A. Wilkinson, IUPAC. Compendium of Chemical Terminology (the “Gold Book”), Blackwell Scientific Publications, Oxford, 2nd edn, 1997 Search PubMed.
- B. Vogelsanger, W. Caminati and A. Bauder, Chem. Phys. Lett., 1987, 141, 245–250 CrossRef CAS.
- S. Ōsawa, M. Sakai and E. Ōsawa, J. Phys. Chem. A, 1997, 101, 1378–1383 CrossRef.
- D. A. Dougherty, C. S. Choi, G. Kaupp, A. B. Buda, J. M. Rudziński and E. Ōsawa, J. Chem. Soc., Perkin Trans. 2, 1986, 1063–1070, 10.1039/p29860001063.
- H. F. Bettinger, P. v R. Schleyer, H. F. Bettinger and H. F. Schaefer III, Chem. Commun., 1998, 769–770, 10.1039/a800741a.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2192929–2192933. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04693h |
| This journal is © The Royal Society of Chemistry 2022 |