
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeted design of porous materials without strong, directional interactions†
Megan
O’Shaughnessy
a,
Peter R.
Spackman
 bcd,
Marc A.
Little
a,
Luca
Catalano
a,
Alex
James
a,
Graeme M.
Day
*b and
Andrew I.
Cooper
*a
bcd,
Marc A.
Little
a,
Luca
Catalano
a,
Alex
James
a,
Graeme M.
Day
*b and
Andrew I.
Cooper
*a
aMaterials Innovation Factory and Department of Chemistry, University of Liverpool, Liverpool, L7 3NY, UK. E-mail: aicooper@liverpool.ac.uk
bComputational System Chemistry, School of Chemistry, University of Southampton, Southampton, SO17 IBJ, UK. E-mail: G.M.Day@soton.ac.uk
cLeverhulme Research Centre for Functional Materials Design, Department of Chemistry and Materials Innovation Factory, University of Liverpool, Liverpool, L7 3NY, UK
dCurtin Institute for Computation, School of Molecular and Life Sciences, Curtin University, PO Box U1987, Perth, Western Australia 6845, Australia
First published on 10th October 2022
Abstract
A porous molecular crystal (TSCl) was found to crystallise from dichloromethane and water during the synthesis of tetrakis(4-sulfophenylmethane). Crystal structure prediction (CSP) rationalises the driving force behind the formation of this porous TSCl phase and the intermolecular interactions that direct its formation. Gas sorption analysis showed that TSCl is permanently porous with selective adsorption of CO2 over N2, H2 and CH4 and a maximum CO2 uptake of 74 cm3 g−1 at 195 K. Calculations revealed that TSCl assembles via a combination of weak hydrogen bonds and strong dispersion interactions. This illustrates that CSP can underpin approaches to crystal engineering that do not involve more intuitive directional interactions, such as hydrogen bonding.
Materials with permanent porosity have widespread applications in areas such as gas storage,1 separation,2 and catalysis.3 Over the past two decades, metal–organic frameworks (MOFs)4 and covalent organic frameworks (COFs)5,6 have emerged as promising classes of porous materials. The strong, directional bonding in MOFs and COFs stabilises the permanent porosity in these materials.7
Porous molecular crystals, by contrast, are solids where the molecular subunits interact via comparatively weak intermolecular interactions.8 These crystals can have either intrinsic or extrinsic porosity. Intrinsic porosity refers to porosity within the molecular subunits, as found in macrocycles9 and porous organic cages (POCs).10 Extrinsic porosity occurs when rigid molecular subunits pack inefficiently, creating pores between the molecules; examples of extrinsically porous molecular materials are hydrogen bonded organic frameworks (HOFs).11 To have true extrinsic porosity, the material must maintain its voids after removal of the guest solvent molecules from the pores.12 This is a challenge for porous molecular crystals, where close packing is typically energetically favoured. Extrinsic permanent porosity results from the interplay between the rigidity and geometrical shape of the molecular building blocks and the strength and directionality of their intermolecular interactions. This is exemplified by Dianin's compound (4-p-hydroxyphenyl-2,2,4-trimethylchroman), which forms inclusion compounds with a range of guest molecules and whose host framework is retained in the absence of guests;13,14 hydroxyl groups form hydrogen-bonded rings which, combined with Dianin's compound's awkward molecular shape, prevent close-packing, leading to the formation of 1-dimensional channels in the host crystal structure. Other classic examples include the porous van der Waals crystals reported by Sozzani and coworkers.15,16
Over the last decade, HOFs have come to be one of the most widely studied classes of porous molecular crystals.17 The most commonly used functional groups in HOFs are 2,4-diaminotriazinyl (DAT) and carboxylic acids due to the strength and directionality of the resulting hydrogen bonds.18 In the quest to design and synthesise novel molecular porous crystals, we turned our attention to a tetrahedral tetraphenylmethane19,20 derivative functionalised with sulfonyl groups (SO2Cl), which have been reported to form cooperative S–Cl⋯O halogen bonding in α,ω-di(sulfonyl chloride) perfluoroalkanes.21 Out aim was to find alternative non-covalent self-assembly motifs for porous molecular crystals.
We report the first example of a porous molecular crystal, TSCl, that uses a sulfonyl chloride as the directing functional group. To do this, the tetrahedral tecton, tetraphenylmethane, was functionalised with sulfonyl chloride groups. CSP has previously been shown to be able to predict the structures of inclusion compounds:22 here by using CSP, we could predict the formation of the porous TSCl structure and, subsequently, use theoretical calculations to understand the molecular interactions in the crystal structure. TSCl crystallises into a network containing open channels directly during its synthesis, and it can be activated readily to exhibit permanent porosity.
TSCl was synthesised by slowly adding an excess of chlorosulfonic acid to tetraphenylmethane in anhydrous dichloromethane (DCM) under an inert atmosphere. The addition of the reaction mixture to ice led to the instant precipitation of TSCl in an 82% yield (see ESI,† Scheme S1). TSCl was isolated in high purity directly as a precipitate from the reaction, as confirmed by 1H and 13C NMR (see ESI,† Fig. S1).
Single crystals of TSCl that were suitable for single crystal X-ray diffraction (SCXRD) were isolated by slowly evaporating 1,1,1,3,3,3-hexafluoro-2-propanol (HIPF), affording single crystals within 3 days. SCXRD reveals that TSCl crystallises in the tetragonal space group I4 with ¼ of the molecule residing in the asymmetric unit. In the crystal structure, each molecule can be viewed as connected to four neighbouring molecules using intermolecular interactions creating a 3D architecture containing tubular channels along the c axis. Using CrystalExplorer model energies,23,24 the intermolecular interactions in TSCl consist of weak hydrogen bonding from the sulfonic oxygens and strong dispersive interactions from the chlorine atoms. There are additional weak hydrogen bonding interactions from the oxygens and hydrogens on the benzene rings. A search of the Cambridge Structural Database (CSD) for the Ph–SO2Cl⋯O motif in TSCl (see ESI,† Fig. S11) gave seven results; all of these structures were densely packed (see ESI,† Fig. S12 and S13). In the seven CSD hits, the most common intermolecular interaction appears to be the halogen bond between the chlorine and oxygen atoms.25–27 For the interaction between the chlorine and oxygen to be classified as halogen bonding, the distances are typically less than the sum of the van der Waals radii,28–30 which is 3.27 Å. To our surprise, in TSCl the distance between the Cl and O is 3.45 Å and 3.70 Å (Fig. 1b), suggesting that the chlorine atom is not involved in any typical halogen bonding interactions.
 | ||
| Fig. 1 (a) Packing of TSCl viewed down the c-axis showing the open accessible voids, (b) the intermolecular interactions in TSCl. | ||
Preliminary rigid-molecule CSP was performed on an ensemble of 17 unique conformers from a gas-phase conformer search to assess the viability of prediction for this molecule. The method used a distributed multipole-based atom–atom force field31 for intermolecular interactions and a quasi-random search for generation of trial crystal structures. Full details, including the generation of conformers and the CSP methodology are provided in the ESI.† The preliminary CSP results on the gas-phase conformer ensemble produced no good match for the experimental TSCl structure. As a test of whether the effect of the polar environment in the crystal on molecular conformations could be significant, CSP was subsequently performed on the ensemble of 17 unique conformers after optimization in a molecular crystal-like dielectric. Likewise, the distributed multipole analysis was performed on the optimised electron density with the dielectric model of the crystalline environment.32
A notable ‘spike’ in the energy versus density distribution of predicted crystal structures was found at a density of approximately 1.3 g cm−3 (Fig. 2a). Such low-density spikes have been found in several cases to correspond to observable porous crystal structures.33,34 This porous crystalline structure (TSCl-1), roughly 10 kJ mol−1 higher in lattice energy than the predicted global minimum energy structure (see ESI,† Fig. S9), was found to reproduce the experimental TSCl crystal structure closely (Fig. 2b). It is worth noting that the molecular geometry in the experimental structure does not correspond to a minimum in the gas-phase—free optimization in the absence of the dielectric environment distorts the molecular geometry significantly—whereas the aforementioned crystal-like dielectric leads to a good match to the molecular conformation in the experimental crystal. This in turn leads to successful reproduction of the experimental crystal structure. Indeed, the set of molecular conformations in the gas-phase are quite distinct from those found when a crystal-like dielectric environment is introduced (see Table S2 in the ESI†). We conclude that the effect of the crystalline environment on molecular structure is important for this structure and, in this case, it is successfully modelled by introducing a continuum dielectric during generation of the conformational ensemble that was input into CSP. The polarizing environment also influences electrostatic interactions, resulting in the observed shift in calculated lattice energies (Fig. 2a) and making the low-density spike more prominent. However, this enhancement of electrostatic interactions seems to be less crucial in reproducing the observed porous crystal structure than the influence of the polarising environment on the molecular geometry.
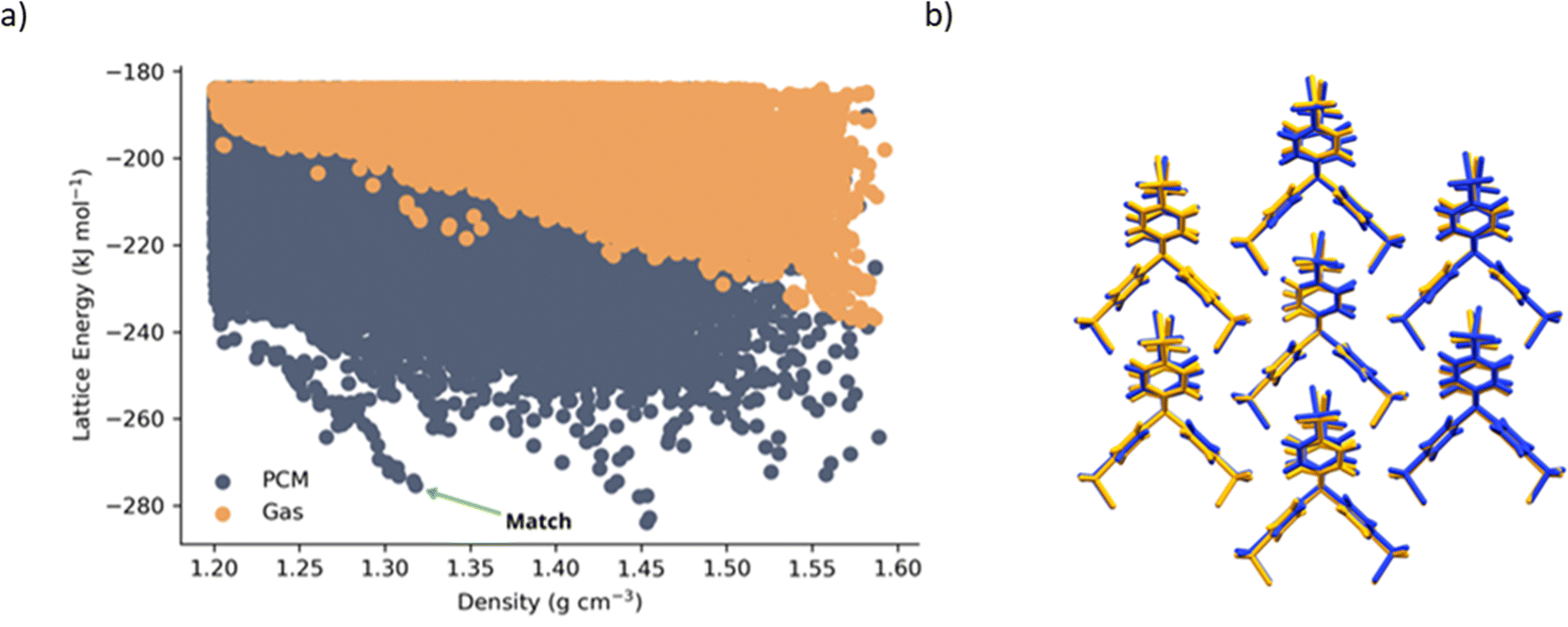 | ||
| Fig. 2 (a) CSP energy-density landscape for TSCl. ‘PCM’ indicates conformers were optimised in a crystal-like dielectric (ε = 4) along with the subsequent distributed multipole analysis, whereas ‘gas’ indicates the usual isolated gas-phase molecular geometries and electron densities were used. (b) Structural overlay of the experimental (orange) and predicted (blue) structure of TSCl, rendered using Mercury35 | ||
Powder X-ray diffraction (PXRD) analysis showed that this porous TSCl phase was formed during the synthesis of tetrakis(4-sulfophenyl)methane and indicated that the crystalline material was phase pure (see ESI,† Fig. S4). Although the porous structure of TSCl was obtained directly from the reaction solvent, attempting to activate it proved difficult because of the large volume of strongly-bound water that was contained within the pores, which was approximately 13 wt%, as revealed by thermogravimetric analysis.
To allow for a simpler activation process that avoids high activation temperatures, TSCl was therefore first recrystallised. The solvents chosen for the recrystallisation were HIFP and methyl acetate (MeOAc). These solvents were selected because of their high vapour pressures at room temperature (20–25 °C), with HIFP having a vapour pressure of 16 kPa and MeOAc 28.8 kPa, compared to 2.4 kPa for water. The addition of MeOAc to HIPF resulted in TSCl instant precipitation with high crystallinity, thus allowing for a fast and simple recrystallisation process. Fig. S6 (ESI†) shows the difference in solvent retention before and after recrystallising TSCl from HIFP/MeOAc, where the weight percentage of solvent has decreased from 25% when isolated straight from the reaction to 7% after the recrystallization.
The recrystallised TSCl material was activated before gas sorption analysis by holding the sample under a nitrogen flow for several hours before being held under a dynamic vacuum at RT on the gas port. The gas sorption isotherms show that TSCl selectively adsorbs CO2 over N2, CH4 and H2 (Fig. 3). The low adsorption of H2 (8 cm3 g−1) is likely due to the weak interaction of the molecules with the structure due to the small kinetic diameter (289 pm) of the H2 molecules allowing the gas to move through the open channels freely. The quadrupole moment of CO2 is approximately three times that of N2,36 which might allow the CO2 molecules to form stronger electrostatic interactions with the sulfonate oxygens of the TSCl structure, providing a possible explanation for the CO2/N2 selectivity. It is also possible that diffusion of N2 is kinetically restricted in this small-pore molecular crystal at 77 K; indeed, the hysteresis in the CO2 isotherm at 195 K also suggests that equilibrium has not been reached, and this hysteresis decreases at higher temperatures. This lack of N2 uptake in molecular crystals with small micropores at low temperatures is relatively common. The TSCl structure had a maximum CO2 uptake of 74 cm3 g−1 at 195K, which is comparable to or greater than other reported porous molecular crystals, including HOF-7a (28 cm3 g−1),37 HOF-3a (∼50 cm3 g−1),38 and HOF-GS-10 (∼42 cm3 g−1),39 all of which also showed selective adsorption of CO2 over N2. PXRD was used to monitor the stability of TSCl to activation and sorption cycles and showed the TSCl remained unchanged (see ESI,† Fig. S9).
In summary, we have found a novel sulfonyl chloride porous molecular crystal, TSCl, which unlike most porous molecular crystals does not pack into a porous structure via strong directional interaction such as hydrogen bonds. Rather, the porous channels in TSCl are a result of the molecular shape of the tetrahedral molecular core, coupled with a combination of weaker intermolecular interactions. TSCl crystals can be grown rapidly from solution and recrystallised within minutes from volatile solvents, which allows for the material to be activated at room temperature under dynamic vacuum. The gas sorption results show that TSCl has permanent porosity, with the structure showing selectivity for CO2 over N2. The material adsorbs a relatively large amount of CO2 compared to other materials in this class, such as HOFs.
We believe this work has implications for future design of porous materials, where the molecular shape of the molecule and the peripheral functional groups are used for the materials design, rather than relying so much on strong directional interactions, such as carboxylic acid dimers, or benzimidazalone groups.40 The use of CSP unlocks this design approach: for example, we do not expect all tetraphenylmethane derivatives to generate porous structures, but chemical knowledge and intuition can fail us in terms of choosing which functionalities to use. CSP methods offer a straightforward way to make this choice – for example, by choosing functionalities that lead to low-energy ‘spikes’ of stable porous structures such as shown in Fig. 2a, or energy landscapes where the global energy minimum is porous, as is the case for Dianin's compound.22 The use of highly directional supramolecular tectons has been successful, but these intuitive design rules are not infallible, and it does limit us to a restricted palette of functionalities, such as carboxylic acids. CSP methods unlock a straightforward method to broaden the range of chemistries that we can consider in supramolecular chemistry. Moreover, the timescales for these CSP calculations using modern methods and hardware are now competitive with the timescale for experimental synthesis and characterization: for example, the data shown in Fig. 2a required approximately 50 thousand CPU hours and could be obtained in around 48 hours on a shared high-performance computing system.
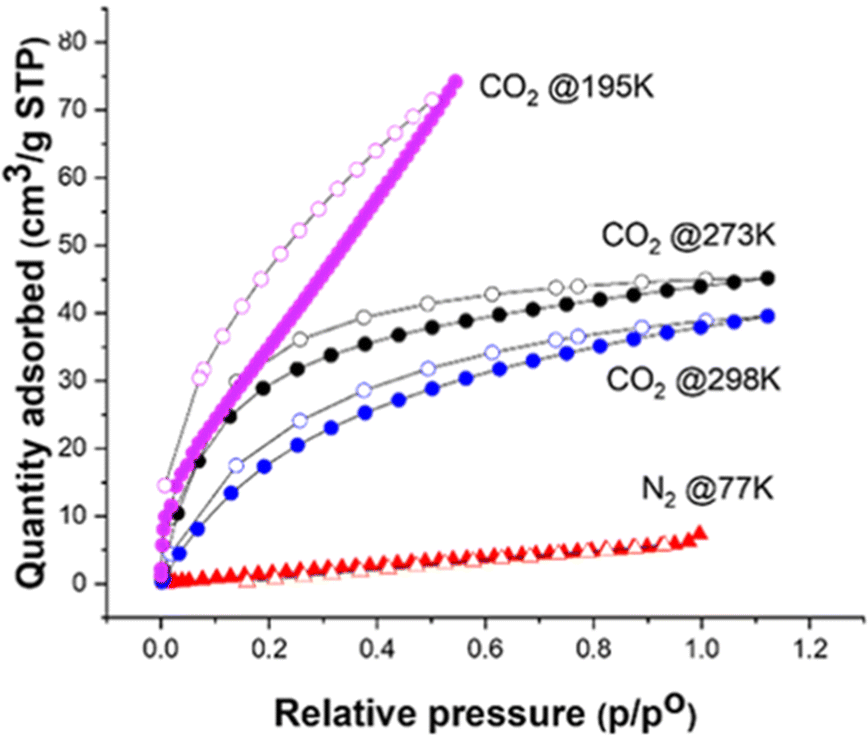 | ||
| Fig. 3 Gas sorptions isotherms for TSCl show selectivity for CO2 over N2. Filled symbols are for adsorption, and the open symbols for desorption. See ESI,† Fig. S8 for H2 and CH4 isotherms. | ||
P. R. S., A. I. C. and G. M. D. would like to acknowledge the Leverhulme centre for functional materials design for funding. We acknowledge the use of the IRIDIS high performance computing facility, and associated support services at the University of Southampton.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, J. Am. Chem. Soc., 2008, 130, 11580–11581 CrossRef CAS PubMed.
- P. Li, Y. He, H. D. Arman, R. Krishna, H. Wang, L. Weng and B. Chen, Chem. Commun., 2014, 50, 13081–13084 RSC.
- Y. Zhang, X. Yang and H.-C. Zhou, Polyhedron, 2018, 154, 189–201 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O’Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15507–15509 CrossRef CAS PubMed.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, 6238 CrossRef.
- S. Dawn, M. B. Dewal, D. Sobransingh, M. C. Paderes, A. C. Wibowo, M. D. Smith, J. A. Krause, P. J. Pellechia and L. S. Shimizu, J. Am. Chem. Soc., 2011, 133, 7025–7032 CrossRef CAS PubMed.
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS.
- J. Luo, J.-W. Wang, J.-H. Zhang, S. Lai and D.-C. Zhong, CrystEngComm, 2018, 20, 5884–5898 RSC.
- L. J. Barbour, Chem. Commun., 2006, 1163–1168 RSC.
- J. L. Flippen, J. Karle and I. L. Karle, J. Am. Chem. Soc., 1970, 92, 3749–3755 CrossRef CAS.
- R. M. Barrer and V. H. Shanson, J. Chem. Soc., Chem. Commun., 1976, 333–334 RSC.
- P. Sozzani, A. Comotti, R. Simonutti, T. Meersmann, J. W. Logan and A. Pines, Angew. Chem., Int. Ed., 2000, 39, 2695–2699 CrossRef CAS.
- P. Sozzani, S. Bracco, A. Comotti, L. Ferretti and R. Simonutti, Angew. Chem., Int. Ed., 2005, 44, 1816–1820 CrossRef CAS.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- J. A. Zerkowski, J. C. MacDonald and G. M. Whitesides, Chem. Mater., 1994, 6, 1250–1257 CrossRef CAS.
- I. Bassanetti, S. Bracco, A. Comotti, M. Negroni, C. Bezuidenhout, S. Canossa, P. P. Mazzeo, L. Marchió and P. Sozzani, J. Mater. Chem. A, 2018, 6, 14231–14239 RSC.
- G. Xing, I. Bassanetti, S. Bracco, M. Negroni, C. Bezuidenhout, T. Ben, P. Sozzani and A. Comotti, Chem. Sci., 2019, 10, 730–736 RSC.
- X. Liu, C. D. McMillen and J. S. Thrasher, New J. Chem., 2018, 42, 10484–10488 RSC.
- A. J. Cruz-Cabeza, G. M. Day and W. Jones, Chem. – Eur. J., 2009, 15, 13033–13040 CrossRef CAS.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- H. Christoph, J. Grunenberg, H. Hopf, I. Dix, P. G. Jones, M. Scholtissek and G. Maier, Chem. – Eur. J., 2008, 14, 5604–5616 CrossRef CAS PubMed.
- S. D. Robertson, A. M. Z. Slawin and J. D. Woollins, Acta Crystallogr., Sect. E, 2006, E62, o744–o745 CrossRef.
- M. Treskow, J. Neudörfl and R. Giernoth, Eur. J. Org. Chem., 2009, 3693–3697 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- L. Brammer, Faraday Discuss., 2017, 203, 485–507 RSC.
- H. Eramian, Y.-H. Tian, Z. Fox, H. Z. Beneberu and M. Kertesz, J. Phys. Chem. A, 2013, 117, 14184–14190 CrossRef CAS.
- S. L. Price, M. Leslie, G. W. A. Welch, M. Habgood, L. S. Price, P. G. Karamertzanis and G. M. Day, Phys. Chem. Chem. Phys., 2010, 12, 8478–8490 RSC.
- T. G. Cooper, K. E. Hejczyk, W. Jones and G. M. Day, J. Chem. Theory Comput., 2008, 4, 1795–1805 CrossRef CAS PubMed.
- A. Pulido, L. J. Chen, T. Kaczorowski, D. Holden, M. A. Little, S. Y. Chong, B. J. Slater, D. P. McMahon, B. Bonillo, C. J. Stackhouse, A. Stephenson, C. M. Kane, R. Clowes, T. Hasell, A. I. Cooper and G. M. Day, Nature, 2017, 543, 657–664 CrossRef CAS PubMed.
- P. Cui, D. P. McMahon, P. R. Spackman, B. M. Alston, M. A. Little, G. M. Day and A. I. Cooper, Chem. Sci., 2019, 43, 9988–9997 RSC.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- C. Graham, J. Pierrus and R. E. Raab, Mol. Phys., 1989, 4, 939–955 CrossRef.
- W. Yang, B. Li, H. Wang, O. Alduhaish, K. Alfooty, M. A. Zayed, P. Li, H. D. Arman and B. Chen, Cryst. Growth Des., 2015, 15, 2000–2004 CrossRef CAS.
- P. Li, Y. He, Y. Zhao, L. Weng, H. Wang, R. Krishna, H. Wu, W. Zhou, M. O’Keeffe, Y. Han and B. Chen, Angew. Chem., Int. Ed., 2014, 574–577 Search PubMed.
- A. Karmakar, R. Illathvalappil, B. Anothumakkool, A. Sen, P. Samanta, A. V. Desai, S. Kurungot and S. K. Ghosh, Angew. Chem., Int. Ed., 2016, 55, 10667–10671 CrossRef CAS.
- M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: For CSP datasets. CCDC 2192388. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04682b |
| This journal is © The Royal Society of Chemistry 2022 |