
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel triple mutant of an extremophilic glycosyl hydrolase enables the rapid synthesis of thioglycosides†
Lauriane
Pillet
a,
David
Lim
a,
Nourah
Almulhim
b,
Ana I.
Benítez-Mateos
 a and
Francesca
Paradisi
*ab
a and
Francesca
Paradisi
*ab
aDepartment of Chemistry, Biochemistry, and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: francesca.paradisi@unibe.ch
bSchool of Chemistry, University of Nottingham, University Park, NG7 2RD, Nottingham, UK
First published on 6th October 2022
Abstract
In order to expand the toolbox of enzymes available for thioglycoside synthesis, we describe here the first example of an extremophilic glycosyl hydrolase from Halothermothrix orenii (HorGH1) engineered towards thioglycosynthase activity with a novel combination of mutations. Using the triple mutant, HorGH1 M299R/E166A/E354G, a range of thioglycosides from glycosyl fluoride donors and aromatic thiols could be synthesised with exquisite stereoselectivity and good to excellent conversions (61–93%).
Over a number of years, the ever-growing interest in biocatalysis for the production of diverse natural products and pharmaceutical ingredients has been firmly established.1 These renewable catalysts not only possess an excellent regio/stereoselectivity, but they also often provide high catalytic efficiencies while operating under much milder conditions.2 In the last decade alone, a myriad of reactions have been reported due to major advances in enzyme engineering, high-throughput screening and process design, which have dramatically expanded the synthetic chemists’ toolbox.1–4 Consequently, biocatalysis can offer several advantages over conventional chemical synthetic methods.1–4
The enzymatic synthesis of thioglycosides is especially underdeveloped despite their presence in many commercial products and pharmaceuticals.5,6 These stable glycosidic analogues confer additional stability to drugs due to their low susceptibility to acid/base and enzyme-mediated hydrolysis.7 Moreover, they are tolerated by many biological systems and have been used as enzyme inhibitors in a range of biochemical studies.6–9 Because of the ubiquity and the importance of glycosidic linkages in biology, thioglycosides have found many applications in carbohydrate-based therapeutics,9 for instance, in the reduction of inflammatory leukocyte accumulation,10 and have even shown promising anti-tumor effects.11
A plethora of chemical syntheses to access thioglycosides have been reported,6,12–16 however, these procedures require multi-step protecting group strategies and offer limited stereochemical control.8 Moreover, they often require pre-activation of the donor, toxic reagents and/or catalysts and are mostly performed in organic solvents.6 An interesting strategy to synthesize aryl 1-thioglycosides from unprotected sugars and corresponding thiols was developed by Tanaka et al. using 2-chloro-1,3-dimethylimidazolinium chloride (DMC) as the dehydrative condensing agent. Although this protecting-group-free synthesis is performed in water, complete stereoselectivity was not guaranteed and the triethylammonium by-products complicated downstream processing.17 Similar methods have been recently reported to give access to aryl-thioglycosides with excellent yields, however they either require strong bases,18 fluorine sources in the presence of organic solvents,19 or thiyl-radicals.20
Enzymatic S-glycoside synthesis offers an attractive alternative to chemical approaches. While natural S-glycosyltransferases have been described in the literature, they require expensive NDP-α-D-sugars as substrates.21,22 A cheaper and attractive method to access these products is therefore desired. In order to address these challenges, enzymatic syntheses using mutant forms of glycosidases have been developed.23–34 Mutant glycosidases which are functional in the synthetic direction –aptly termed glycosynthases or glycoligases- are derived from glycosyl hydrolases which have had their catalytic nucleophile and/or acid/base residue replaced with non-nucleophilic residues, such as glycine or alanine. Glycosynthases are inherently unable of hydrolyzing their natural substrates. Nevertheless, when supplemented with an activated donor, such as a glycosyl fluoride, transglycosylation may occur without subsequent hydrolysis of the product due to the nature of the introduced mutation. While thiooligosaccharides have been synthetized with thioglycoligases23–25 (single acid/base mutant) and with thioglycosynthases26,27 (double acid/base and nucleophilic mutant), enzymatic syntheses of thioglycoconjugates are scarce.28–30 Moreover, attempts to translate these elegant proof-of-concepts to industrial processes are often plagued by a very low concentration of reactants1–4 and enzymes being poorly resistant to the organic co-solvents that are often required to dissolve thiols.1–3
In order to expand the range of reaction conditions suitable for biocatalytic preparations of thioglycosides, we selected a β-glycosyl hydrolase (GH1; EC 3.2.1.21) from Halothermothrix orenii (HorGH1) for further investigation.31 This enzyme has been shown to perform well under non-physiological conditions.32,33 Enzymes from extremophilic organisms indeed possess a significantly higher tolerance to extreme temperatures, pHs and organic cosolvents compared to their mesophilic counterparts.34,35
We previously reported on the engineering of a β-glycosidase into a β-thioglycosidase via rational design and highlighted the key role of an arginine residue in the recognition of thioglycosides. The HorGH1 M299R mutant exhibited a 3-fold increase in specificity for the hydrolysis of β-thioglycosides without any loss in turnover rate compared to the wild type (WT) enzymes.36
The depletion of catalytic residues26,27 combined with the HorGH1 M299R mutant (Fig. S1, ESI†) could yield a very powerful catalyst for the synthesis of still elusive thioglycosides. To date, single acid/base mutants of a GH20 hexosaminidase,28 a GH3 β-glycosidase29 and of a thermophilic glucosidase30 have shown to be able to access thioglycoconjugates, but a double deletion in any enzyme for this reaction has not been reported. Here, the mutations E166A (acid/base residue) and E354G (nucleophilic residue) were introduced into the catalytic site of HorGH1 WT and HorGH1 M299R as single, double and triple mutants (generating 8 HorGH1 variants, Table 1) in order to understand which combination leads to the most efficient thioglycosynthase. Herein, we present the first example of an extremophilic glycosyl hydrolase from the GH1 family engineered towards thioglycosynthase activity and shed light on the differences between the thioglycoligase and the thioglycosynthase strategies in a same enzyme.
| Type of glycosidase | Catalytic residues maintained | Acid/base residue removed | Nucleophilic residue removed | Nucleophilic and acid/base residue removed |
|---|---|---|---|---|
| Reactions were performed in the presence of 1 mM sugar donor, 10 mM thiol acceptor at 25 °C in bicarbonate buffer pH 9.4 containing 30% DMSO and the enzyme (1 mg mL−1). Reactions were all run for 24 h. Conversions were determined by HPLC analysis based on product formation. | ||||
| β-Glycosidase | Wild type | E166A | E354G | E166A/E354G |
| β-Thioglycosidase | M299R | M299R/E166A | M299R/E354G | M299R/E166A/E354G |
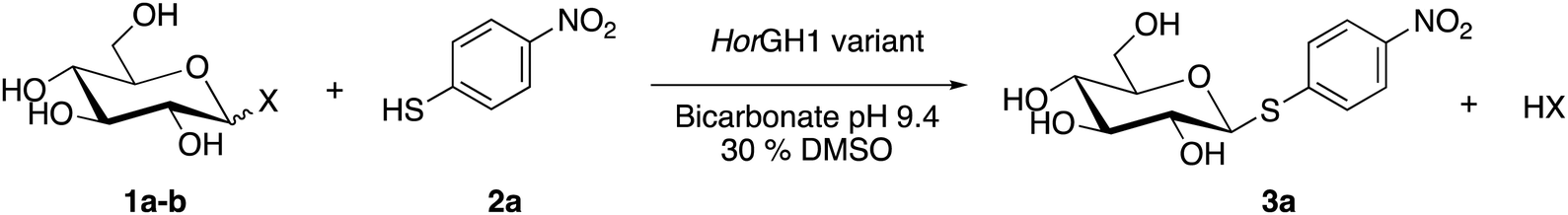
All mutants were successfully obtained in pure form (Fig. S2, ESI†) and tested for thioglycoside synthesis. Reactions with the variants depleted of the acid/base residue (E166A or E166A/M299R) were performed with p-nitrophenyl-β-D-glucopyranoside (1) as the sugar donor bearing a standard leaving group in the β position (p-nitrophenol (pNP), Table 2 entries 1–4, 1a) and p-nitrothiophenol (2a) as the thiol acceptor. Reactions with the variants lacking the nucleophilic residue (E354G and E354G/M299R) or both acid/base and nucleophilic amino acids (E166A/E354G and E166A/E354G/M299R) were performed using α-D-glucopyranosyl fluoride as the donor instead (F in α, Table 2 entries 5–8, 1b). A good leaving group in the α position is needed with these latter variants to compensate for the absence of the glycosyl–enzyme intermediate normally formed with the nucleophilic residue. In order to alleviate thiol oxidation, the buffers were degassed and various reducing agents were screened. While tris(2-carboxyethyl)phosphine (TCEP) completely inactivates the enzyme at concentrations above 2 mM, and 1,4-dithiothreitol (DTT) reduced conversion by half at a concentration of 20 mM, β-mercaptoethanol was found to be tolerated by the enzyme and was used for further experiments (Fig. S4, ESI†). No formation of thioglycosides was observed upon incubation of the WT enzyme and M299R mutant (Table 2, entries 1 and 2). Moreover, no spontaneous reactions (i.e. direct SN2 displacement of the anomeric group) were observed in the absence of enzyme under our working conditions. The neutralization of the acid/base residue in E166A and M299R/E166A variants (Table 2, entries 3 and 4) induced some thioglycosynthetic capability, but yields were low (20%). This inefficient transglycosylation is likely due to the poor leaving group ability of the p-nitrophenyl substituent in the absence of acid catalysis (Fig. S5, ESI†). Interestingly, no thioglycoside activity was observed with the mutants E354G and M299R/E3354G lacking the nucleophilic residue (Table 2, entries 5 and 6). The variants E166A/E354G and M299R/E166A/E354G depleted of both catalytic residues (Table 2, entries 7 and 8), gave very good (61%) to excellent (92%) conversions with p-nitrothiophenol as acceptor, respectively (Fig. S6, ESI†). The synthetic capability of the variants was significantly enhanced by the M299R mutation and confirms the key role of arginine in the recognition of sulfur atom. The high nucleophilicity of the thiolate tested with the double mutant scaffold stabilizing the transition states,26,27 is indeed sufficient to achieve a significant product yield, but the active site is clearly positively impacted by the M299R mutation, as it was reported for the hydrolysis of thioglycosides.36 Whereas the KM towards α-D-glucopyranosyl fluoride was found to be similar for the M299R/E166A/E354G and the E166A/E354G variants (2.26 and 2.12 mM, respectively), the KM towards pNT was lower for M299R/E166A/E354G (3.82 mM) than for E166A/E354G (7.46 mM) (Fig. S7, ESI†). Analysis of the product by 1H NMR confirmed the presence of a β-thioglycosidic linkage, thereby showing the absolute stereoselectivity of the enzyme (Fig. S8, ESI†). Synthesis of thioglycoside products was also confirmed by HRMS (Table S2, ESI†). As expected, none of the mutants showed any significant hydrolytic activity towards p-nitrophenyl-β-D-glucopyranoside and p-nitrophenyl-β-D-thioglucopyranoside substrates (Fig. S9, ESI†).
|
|
|||
|---|---|---|---|
| Entry | Sugar donor | Enzyme variant | Molar conversion [%] |
| 1 | 1a, X = pNP in β | WT | 0 |
| 2 | 1a, X = pNP in β | M299R | 0 |
| 3 | 1a, X = pNP in β | E166A | 23 |
| 4 | 1a, X = pNP in β | M299R/E166A | 26 |
| 5 | 1b, X = F in α | E354G | 0 |
| 6 | 1b, X = F in α | M299R/E354G | 0 |
| 7 | 1b, X = F in α | E166A/E354G | 61 |
| 8 | 1b, X = F in α | M299R/E166A/E354G | 92 |
The encouraging results obtained with HorGH1 M299R/E166A/E354G variant triggered the exploration of additional thiols that could be accepted by this mutant (Fig. S10, ESI†). The triple mutant (1 mg mL−1) was successfully tested with a range of substituted thiophenols at 1 mM scale. We also observed a pH dependence of the reaction, with pH 9.4 furnishing the highest conversion for all respective substrates (Fig. S11, ESI†). While most of the tested aromatic thiols yielded the corresponding thioglycosides in good to excellent conversions (61–93%, Table 3, entries 1–4 and 6), no conversion was observed with p-aminothiophenol (2e), as well as with N,N′-dibenzoyl-L-cystine and diphenyl diselenide as acceptors (Table 3, entries 7 and 8, respectively). Two additional substrates, p-methoxythiophenol and 2-naphthalenethiol were tried but gave inconsistent results due to poor solubility.
|
|
||||
|---|---|---|---|---|
| Entry | Sugar donor | Acceptor | Reaction time [h] | Molar conversion [%] |
| Reactions were performed in the presence of 1 mM sugar donor, 10 mM thiol acceptor at 25 °C in 30% DMSO, 50 mM bicarbonate buffer pH 9.4, 20 mM of β-mercaptoethanol and the enzyme (1 mg mL−1) for products 3a–d. Conversions were determined by HPLC based on product formation. Reactions were performed in the presence of 10 mM sugar donor, 100 mM thiol acceptor at 25 °C in 30% deuterated DMSO, 50 mM bicarbonate buffer pH 9.4 in D2O, 200 mM β-mercaptoethanol and the enzyme (1 mg mL−1 in D2O) for products 3e–f. Conversion was determined by NMR. | ||||
| 1 | α-D-Glucopyranosyl fluoride (1a) | p-Nitrothiophenol (Y = C, X = NO2, 2a) | 2 | 92 |
| 2 | α-D-Glucopyranosyl fluoride (1a) | Thiophenol (Y = C, X = H, 2b) | 48 | 61 |
| 3 | α-D-Glucopyranosyl fluoride (1a) | p-Thiotoluene (Y = C, X = CH3, 2c) | 48 | 81 |
| 4 | α-D-Glucopyranosyl fluoride (1a) | p-Bromothiophenol (Y = C, X = Br, 2d) | 2 | 93 |
| 5 | α-D-Glucopyranosyl fluoride (1a) | p-Aminothiophenol (Y = C, X = NH2, 2e) | 72 | 0 |
| 6 | α-D-Glucopyranosyl fluoride (1a) | 2-Mercaptopyridine (Y = N, X = H, 2f) | 72 | 70 |
| 7 | α-D-Glucopyranosyl fluoride (1a) | N,N′-Dibenzoyl-L-cystine | 48 | 0 |
| 8 | α-D-Glucopyranosyl fluoride (1a) | Diphenyl diselenide | 48 | 0 |

Besides being easy to handle and cofactor-independent, this robust catalyst remained active for 48 hours despite the presence of 30% DMSO. With an excess of thiol acceptor, cheap starting materials were converted into valuable chemical products with high added value. Moreover, the products formed are interesting intermediates in the synthesis of pharmaceuticals; 3b, for example, in its peracetylated version was shown to efficiently reduce inflammatory leukocyte accumulation by functioning as metabolic decoy,10 while the nitrosylated 3f showed an antitumor effect which was significantly enhanced in combination with insulin.11 Similarly, the bromide substituted 3d could be further derivatized via cross-coupling methodologies for the formation of valuable thioglycoconjuguates.28,37,38 More importantly, the triple mutant approach could be extended to other enzymes that have a different acceptor tolerance.
In conclusion, this work provides critical insights on the differences between the use of thioglycoligases and the thioglycosynthases for the synthesis of thioglycosides in a same thermophilic β-glycosyl hydrolase from the GH1 family. For the first time, mutations of the acid/base (E166A) and the nucleophilic catalytic residues (E354G) into neutral amino acids were combined with a thiol-specific mutation (M299R) aiming to increase specificity towards β-thioglycoside synthesis. The repertoire of mutant glycosidases available for the biosynthesis of thioglycosides was expanded by constructing an evolved enzyme, HorGH1 M299R/E166A/E354G, which proved to be an efficient thioglycosynthase as highlighted by the range of accepted aromatic thiols. The exquisite stereoselectivity and the good yields obtained demonstrated the catalytic efficiency of this enzyme. Our engineered enzyme provides an innovative, safe, green and profitable synthetic route for the construction of S-glycosidic linkages. By endowing robust and efficient enzymes with new functions, we can pave the way to the effective implementation of biocatalytic processes at the industrial scale while alleviating their ecological imbalance.
F. P. conceptualized the idea and supervised the project. L. P. performed most of the experimental work and wrote the initial draft. D. L. guided the biotransformations and synthesized all standards. N. A. developed the double mutated versions of the enzyme. A. I. B.-M. designed and supervised the genetic experiments. All authors discussed and agreed to the final version of the manuscript.
This project was supported by the SNSF (200021_192274, F.P.) and the University of Bern “Seal of Excellence Fund” Postdoctoral Fellowship (SELF19-03 BIORPHANDRUG, A. I. B.-M) for funding this research. Open access funding was provided by Universität Bern.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- J. M. Woodley, Philos. Trans. R. Soc., A, 2018, 376(2110), 20170062 CrossRef PubMed.
- R. A. Rocha, R. E. Speight and C. Scott., Org. Process Res. Dev., 2022, 26(7), 1914–1924 CrossRef CAS.
- D. C. Miller, S. V. Athavale and F. H. Arnold, Nat. Synth., 2022, 1(1), 18–23 CrossRef PubMed.
- M. L. Contente, D. Roura Padrosa, F. Molinari and F. Paradisi, Nat. Catal., 2020, 3, 1020–1026 CrossRef CAS.
- M. Qiao, L. Zhang, R. Jiao, S. Zhang, B. Li and X. Zhang, Tetrahedron, 2021, 81, 131920 CrossRef CAS.
- K. Pachamuthu and R. R. Schmidt, Chem. Rev., 2006, 106(1), 160–187 CrossRef CAS PubMed.
- N. Agerbirk and C. E. Olsen, Phytochemistry, 2012, 77, 16–45 CrossRef CAS PubMed.
- Y. W. Kim, H. Chen, J. H. Kim and S. G. Withers, FEBS Lett., 2006, 580(18), 4377–4381 CrossRef CAS PubMed.
- F. Castaneda, A. Burse, W. Boland and R. K. Kinne, Int. J. Med. Sci., 2007, 4(3), 131–139 CrossRef CAS PubMed.
- S. S. Wang, X. Gao, V. Del Solar, Y. Xinheng, A. Antonopoulos, A. E. Friedman, E. K. Matich, G. E. Atilla-Gokcumen, M. Nasirikenari, J. T. Lau, A. Dell, S. M. Haslam, R. A. Laine, K. L. Matta and S. Neelamegham, Cell Chem. Biol., 2018, 25(12), 1519–1532.e5 CrossRef CAS PubMed.
- S. Agrawal, M. Wozniak, M. Luc, K. Walaszek, E. Pielka, W. Szeja, G. Pastuch-Gawolek, A. Gamian and P. Ziolkowski, Oncotarget, 2017, 8, 114173 CrossRef PubMed.
- S. Escopy and A. V. Demchenko, Chem. – Eur. J., 2022, 28(14), e202103747 CAS.
- G. Hummel and O. Hindsgaul, Angew. Chem., Int. Ed., 1999, 38(12), 1782–1784 CrossRef CAS PubMed.
- B. D. Johnston and B. M. Pinto, J. Org. Chem., 2000, 65(15), 4607–4617 CrossRef CAS PubMed.
- S. K. Das, J. Roy, K. A. Reddy and C. A. Abbineni, Carbohydr. Res., 2003, 338(21), 2237–2240 CrossRef CAS.
- C. A. Tai, S. S. Kulkarni and S. C. Hung, J. Org. Chem., 2003, 68(22), 8719–8722 CrossRef CAS PubMed.
- T. Tanaka, T. Matsumoto, M. Noguchi, A. Kobayashi and S. Shoda, Chem. Lett., 2009, 38(5), 458–459 CrossRef CAS.
- G. L. Zhan, M. R. Gadi, X. Cui, D. Liu, J. Zhang, V. Saikam, C. Gibbons, P. G. Wang and L. Li, Green Chem., 2021, 23(8), 2907–2912 RSC.
- Y. Liu, X. B. Yu, X. M. Zang, Q. Zhong, L. H. Liao and N. Yan, RSC Adv., 2021, 11, 26666–26671 RSC.
- L. McSweeney, F. Dénès and E. M. Scanlan, Eur. J. Org. Chem., 2016,(12), 2080–2095 CrossRef CAS.
- J. Ati, P. Lafite and R. Daniellou, Beilstein J. Org. Chem., 2017, 13, 1857–1865 CrossRef CAS PubMed.
- J. R. Rich, A. Szpacenko, M. M. Palcic and D. R. Bundle, Angew. Chem., Int. Ed., 2004, 43(5), 613–615 CrossRef CAS PubMed.
- M. Jahn, J. Marles, R. A. Warren and S. G. Withers, Angew. Chem., Int. Ed., 2003, 42(3), 352–354 CrossRef CAS PubMed.
- Y. W. Kim, H. M. Chen, J. H. Kim, J. Müllegger, D. Mahuran and S. G. Withers, ChemBioChem, 2007, 8(13), 1495–1499 CrossRef CAS PubMed.
- J. Müllegger, H. M. Chen, W. Y. Chan, S. P. Reid, M. Jahn, R. A. J. Warren, H. M. Salleh and S. G. Withers, ChemBioChem, 2006, 7(7), 1028–1030 CrossRef PubMed.
- S. G. Withers and M. Jahn, World Pat., WO2005/040371, World Intellectual Property Organisation, 2005 Search PubMed.
- M. Jahn, H. Chen, J. Mullegger, J. Marles, R. A. Warren and S. G. Withers, Chem. Commun., 2004, 274–275 RSC.
- G. Tegl, J. Hanson, H. M. Chen, D. H. Kwan, A. G. Santana and S. G. Withers, Angew. Chem., Int. Ed., 2019, 58(6), 1632–1637 CrossRef CAS PubMed.
- M. Nieto-Domínguez, B. Fernández de Toro, L. I. de Eugenio, A. G. Santana, L. Bejarano-Muñoz, Z. Armostrong, J. A. Méndez-Líter, J. L. Asensio, A. Prieto, S. G. Whithers, F. J. Cañada and M. Jesús Martínez, Nat. Commun., 2020, 11(1), 4864 CrossRef PubMed.
- C. Peyrot, B. Didak, L. Guillotin, L. Landemarre, P. Lafite, L. Lemiègre and R. Daniellou, Eur. J. Org. Chem., 2021,(27), 3812 CrossRef CAS.
- L. D. Kori, A. Hofmann and B. K. Patel, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2011, 67(1), 111–113 CrossRef CAS PubMed.
- L. Delgado, M. Parker, I. Fisk and F. Paradisi, Food Chem., 2020, 323, 126825 CrossRef CAS PubMed.
- N. Hassan, T. H. Nguyen, M. Intanon, L. D. Kori, B. K. C. Patel, D. Haltrich, C. Divne and T. C. Tan, Appl. Microbiol. Biotechnol., 2015, 99, 1731–1744 CrossRef CAS PubMed.
- J. Yin, J. C. Chen, Q. Wu and G. Q. Chen, Biotechnol. Adv., 2015, 33(7), 1433–1442 CrossRef CAS PubMed.
- S. Park, B. Lee and K. Park, J. Nutr. Health Food Eng., 2017, 7(1), 230–237 Search PubMed.
- N. Almulhim, N. R. Moody and F. Paradisi, Appl. Microbiol. Biotechnol., 2020, 104(10), 4407–4415 CrossRef CAS PubMed.
- J. Müllegger J, H. M. Chen, R. A. Warren and S. G. Withers, Angew. Chem., Int. Ed., 2006, 45(16), 2585–2588 CrossRef PubMed.
- I. Compañón, A. Guerreiro, V. Mangini., J. Castro-López, M. Escudero-Casao, A. Avenoza, J. H. Busto, S. Castillón, J. Jiménez-Barbero, J. L. Asensio, G. Jiménez-Osés, O. Boutureira, J. M. Peregrina, R. Hurtado-Guerrero, R. Fiammengo, G. J. L. Bernardes and F. Corzana, J. Am. Chem. Soc., 2019, 141(9), 4063–4072 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc04660a |
| This journal is © The Royal Society of Chemistry 2022 |