
Unique regioselectivity of the Pd-catalysed cross-dehydrogenative coupling reaction of simple polyaromatic hydrocarbons with polyfluoroarenes†
Ryota
Sato
,
Tomoki
Iida
,
Takaki
Kanbara
 * and
Junpei
Kuwabara
*
* and
Junpei
Kuwabara
*
Tsukuba Research Center for Energy Materials Science (TREMS) Graduate School of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8573, Japan. E-mail: kanbara@ims.tsukuba.ac.jp; kuwabara@ims.tsukuba.ac.jp
First published on 14th September 2022
Abstract
A Pd-catalysed cross-dehydrogenative coupling reaction introduced two polyfluoroarenes into simple polyaromatic hydrocarbons at sterically favourable positions. An investigation of the reaction mechanism revealed that the unique regioselectivity was determined by the reductive elimination step rather than the C–H bond cleavage step.
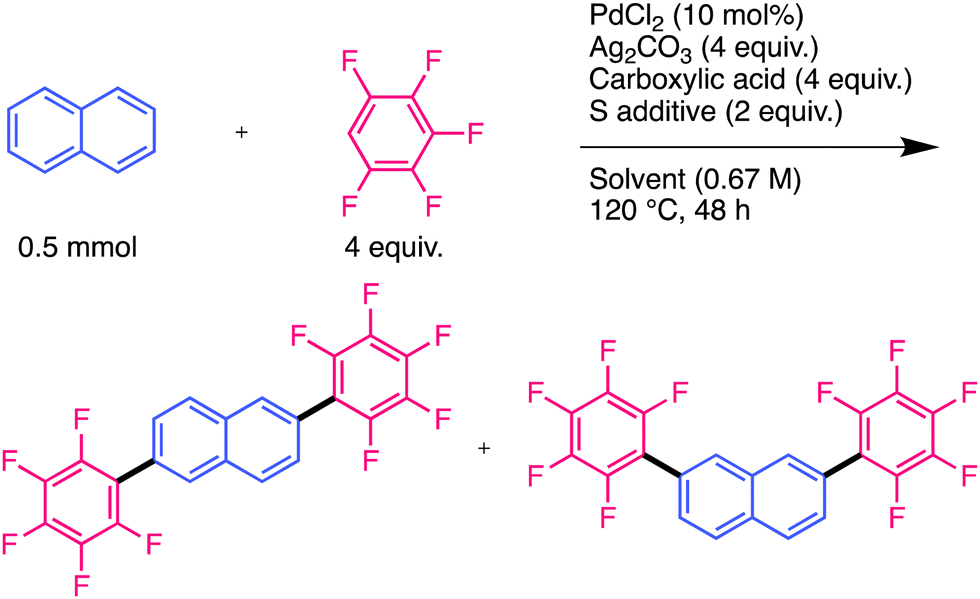
Cross-coupling reactions are promising pathways for biaryl construction and have contributed to the efficient synthesis of organic electronic materials and pharmaceuticals.1–3 Recently, direct C–H arylation reactions have been developed as efficient cross-coupling reactions.4–6 The cross-dehydrogenative coupling (CDC) reaction is the most promising method for synthesising biaryls because it avoids the pre-functionalisation of both aromatic compounds. Since the CDC reaction of indole with benzene was reported by Fagnou et al.,7 efficient CDC reactions of a variety of substrates have been developed in recent years.8,9 Polyfluoroarenes are promising substrates for CDC reactions because of their highly reactive C–H bonds. Moreover, polyfluoroarenes serve as strong electron-deficient functional groups that impart n-type semiconducting properties to aromatic molecules as Yamashita and Hu demonstrated using oligothiophenes and anthracenes.10,11 As anthracene derivatives with two aromatic substituents serve as efficient materials for organic light-emitting diodes (OLEDs) and organic field-effect transistors (OFETs),11–14 it would be beneficial to introduce two aryl groups to anthracene in one step via a CDC reaction. A few studies have demonstrated CDC reactions of polyfluoroarenes with simple aromatic hydrocarbons, such as benzene. Su et al. developed a Pd-catalysed CDC reaction of benzene with polyfluoroarenes using Cu salt as an oxidant (Fig. 1(a)-1).15
 | ||
| Fig. 1 CDC reactions of simple arenes with polyfluoroarenes. | ||
Shi et al. developed a similar reaction using a Pd catalyst; Ag salt was used as the oxidant, and a bulky diisopropyl sulfide was used as an additive (Fig. 1(a)-2).16 Recent studies suggest that Ag salts participate in cleaving the C–H bonds of polyfluoroarenes in addition to their role as oxidants.17–21 However, introducing multiple polyfluoroarenes onto benzene using these methods is challenging because they require excess benzene. Our group demonstrated that a Pd mono-polyfluoroaryl complex acts as a catalytically active species in the CDC reaction of thiophenes with polyfluoroarenes,18 based on a previous study by Zhang et al.20 Furthermore, we found that the bis-polyfluoroaryl Pd resting-state species (RSS) does not cause reductive elimination; instead, it reacts with the Ag complex to generate the catalytically active species (Fig. 1(b)). This unique reactivity causes an efficient cross-coupling reaction without homocoupling side reactions. We hypothesised that multiple polyfluoroarenes could be efficiently introduced onto polyaromatic hydrocarbons (PAHs) using this reaction via highly active mono-polyfluoroaryl Pd intermediates without homocoupling side reactions (Fig. 1(c)). However, because simple PAHs are less reactive than thiophenes,21 this reaction may be very slow. To overcome this limitation, we optimised the reaction conditions to suppress RSS formation, allowing the introduction of multiple polyfluoroarenes onto simple PAHs. Significantly, this reaction preferentially occurred at the sterically vacant positions of PAHs, which is contrary to previous examples and predictions based on calculations.22–25 Mechanistic analysis revealed that this unique regioselectivity was determined by the reductive elimination step rather than the C–H bond cleavage step.
First, we optimised the reaction conditions by focusing on the carboxylic acids, sulfur additives, and solvents (Table 1). The yield of the disubstituted products was low under the conditions described in a previous report on thiophene substrates (entry 1).18 A mixture of 2,6- and 2,7-substituted compounds was produced, as described below. We used 1-adamantanecarboxylic acid, which is bulkier than pivalic acid, and the yield increased slightly (entry 2).26,27 As RSS predominantly forms under high-polarity conditions,18 low-polarity additives and solvents were tested to suppress RSS formation. The use of sulfoxide additive with long alkyl chains improved the yield (entry 3). In addition, a higher yield was obtained when a low-polarity solvent, cyclopentyl methyl ether (CPME), was used instead of N,N-dimethylformamide (DMF) (entry 4). In this reaction, most of the naphthalene was consumed, but the monosubstituted compound remained in the crude products (Table S1, ESI†). These results indicated that lower polarity conditions are effective for introducing multiple polyfluoroarenes into simple PAHs with low reactivity. Disubstituted products were obtained in 64% yield using 2,3,5,6-heptafluorotoluene (HFT) as the substrate (entry 5). To reliably determine the regioselectivity of these reactions, authentic samples of 2,6- and 2,7-substituted isomers were synthesised via a direct arylation reaction (see ESI†). The structure of the 2,6-isomer was confirmed using single-crystal X-ray structural analysis (Fig. S1a, ESI†). Comparison with the NMR spectra of authentic samples confirmed that the products of the current CDC reaction are 2,6- and 2,7-isomers. We separated the isomers by exploiting the difference in their solubility; the 2,6-isomer has a lower solubility in common organic solvents than the 2,7-isomer. Detailed NMR spectrum assignments and isolation experiments showed that the yield of the 2,7-isomer was higher than that of the 2,6-isomer (Table 1, entries 4 and 5).
|
|
||||
|---|---|---|---|---|
| Entry | RCOOH | S additive | Solvent | Yielda (%) |
| a Yields of 2,6- and 2,7-substituted compounds were determined using 19F NMR analysis of the crude product with hexafluorobenzene as an internal standard. b Use of 2,3,5,6-heptafluorotoluene instead of pentafluorobenzene. c Ratio of the isolated 2,6- and 2,7-isomers. d Yield of a mixture of bis-((2,3,5,6-tetrafluoro-4-trifluoromethyl)phenyl)naphthalenes after purification using column chromatography. e Ratio of 2,6- and 2,7-isomers obtained from the 1H NMR spectrum of the mixture. | ||||
| 1 | PivOH | DMSO | DMF | 2 |
| 2 | 1-AdCOOH | DMSO | DMF | 7 |
| 3 | 1-AdCOOH | Octyl2SO | DMF | 22 |
| 4 | 1-AdCOOH | Octyl2SO | CPME | 37 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.4)c :2.4)c |
| 5b | 1-AdCOOH | Octyl2SO | CPME | 64d (1:3.2)e |
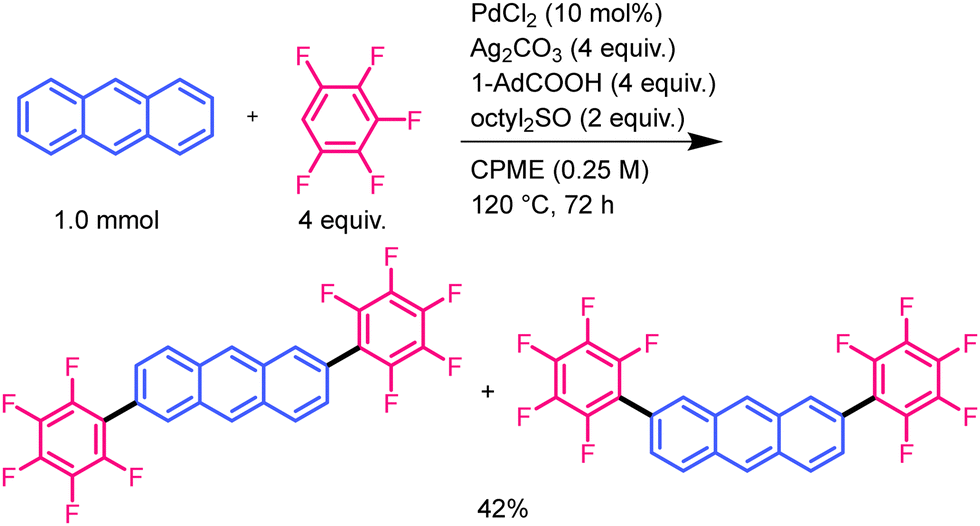
The reaction of anthracene with pentafluorobenzene was also performed under the same conditions to confirm its generality and regioselectivity (Scheme 1). This reaction proceeded in a regiospecific manner and produced a mixture of 2,6- and 2,7-bis(pentafluorophenyl)anthracenes in 42% yield. The 2,6- and 2,7-isomers were isolated from the mixture via solvent washing with 9.2% and 10.6% yields, respectively. The isomers were identified by comparison with authentic samples. The structure of 2,6-bis(pentafluorophenyl)anthracene was identified using single-crystal X-ray structural analysis (Fig. S1b, ESI†).
 | ||
| Scheme 1 CDC reaction of an anthracene with two pentafluorobenzenes. | ||
In both cases, the reactions occurred at the 2,6- and 2,7-positions. Such β-selectivity is rare in C–H bond functionalisation using Pd catalysts,6,24 as most Pd-catalysed C–H bond cleavages proceed via the concerted metalation-deprotonation (CMD) pathway.22,28–30 Density functional theory calculations for the CMD pathway predict that the reaction is more likely to proceed at the α-position of naphthalene than at the β-position,31 and experimental results supported the prediction.22,24,25,28–31 Sanford et al. also reported the α-selective C–H bond functionalisation of naphthalene using a Pd catalyst while β-selective reaction required a Pt(IV) catalyst.25 To identify the reason for the unique regioselectivity of this reaction, the reaction mechanism was investigated.
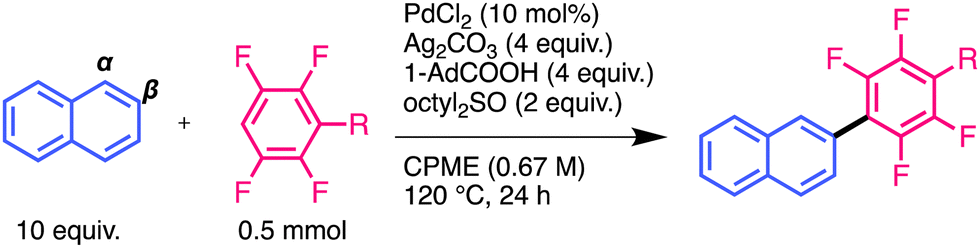
First, we investigated the monosubstitution reactions of naphthalene with polyfluoroarenes (Table 2). β-pentafluorophenylnaphthalene was obtained in a high yield, as in the case of the disubstituted products (entry 1). Moreover, the reactions of polyfluoroarenes bearing an electron-donating OMe group or an electron-withdrawing CF3 group generated high yields of β-substituted compounds (entries 2 and 3). These results indicated that the substituents of polyfluoroarenes at the para-position had no effect on β-selectivity.
Second, we focused on naphthalene to determine the origin of its selectivity. Deuteration experiments using naphthalene-d8 were performed to investigate the reversibility of the C–H bond cleavage (Fig. 2). The 1H NMR spectrum of the monosubstituted product showed signals attributed to the α (1, 4, 5, 8) position. Deuterium at the α-position was exchanged with protons, indicating reversible C–H bond cleavage. The proton source was confirmed to be 1-adamantanecarboxylic acid via another deuterium reaction (Fig. S2, ESI†). In contrast, no proton signals corresponding to the β (3, 6, 7) positions were observed (Fig. 2 and Fig. S2, ESI†). This result suggested that the reverse reaction of C–H bond cleavage does not occur at the β-position, presumably because of rapid reductive elimination. The reaction mechanism was estimated based on the experimental results, as shown in Scheme 2.
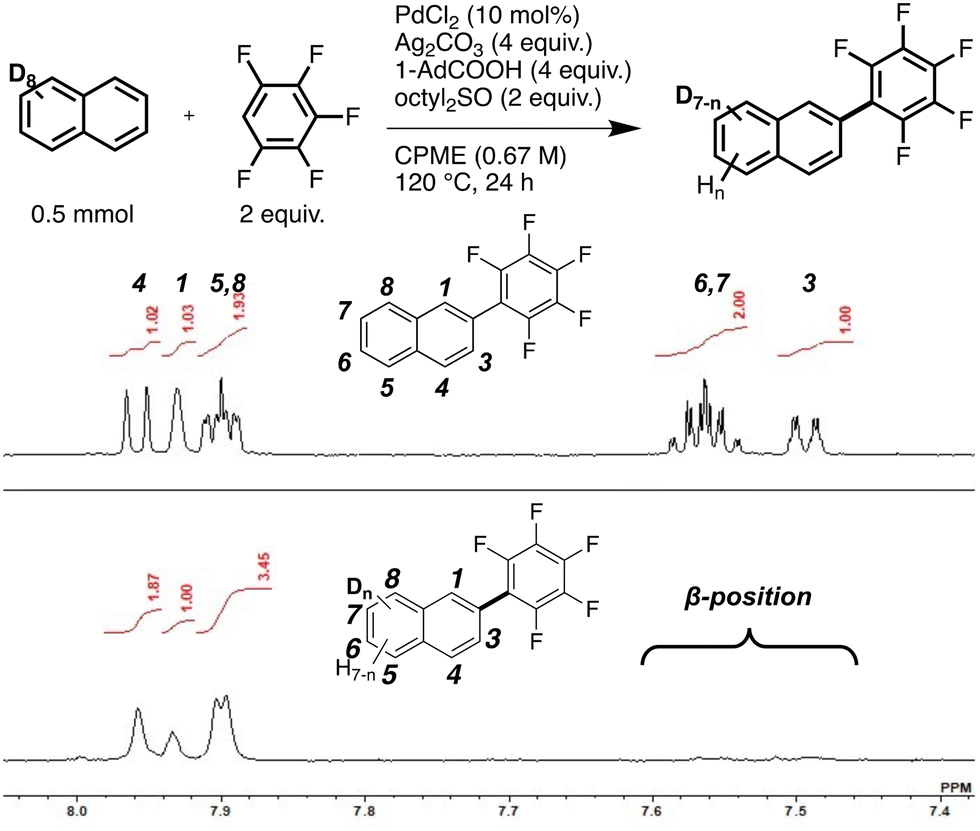 | ||
| Fig. 2 Reaction scheme; 1H NMR spectra of 2-pentafluorophenylnaphthalene (top) and the product (bottom) (600 MHz, CDCl3, r.t.). | ||
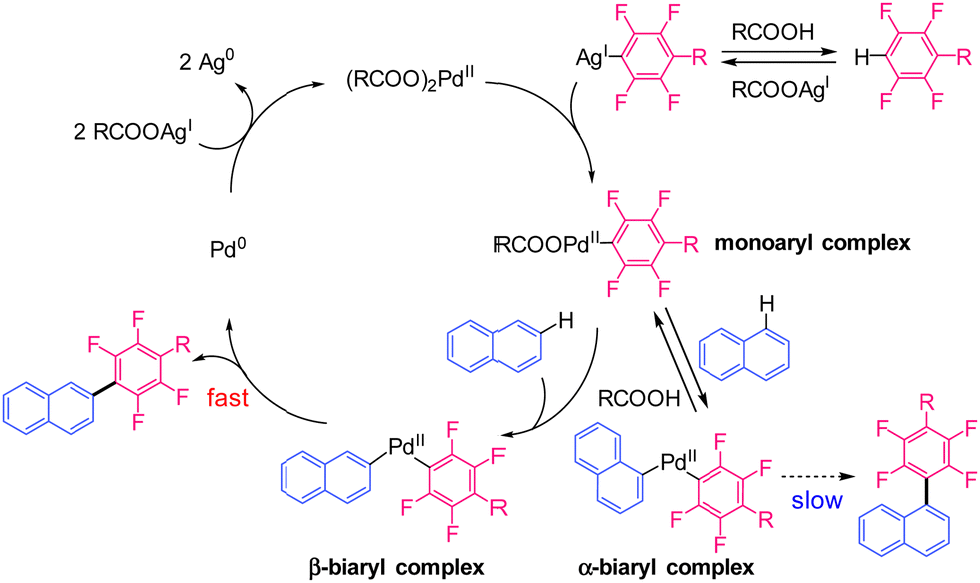 | ||
| Scheme 2 Possible reaction mechanism. | ||
The monoaryl Pd complex is generated by the transmetallation of Pd(OOCR)2 with the Ag-polyfluoroaryl complex. This species reacts with the C–H bond of naphthalene at the α- or β-positions. The reductive elimination of the α-biaryl Pd complex is likely to be very slow because of the large steric hindrance at the α-position and the strong electron-withdrawing effect of the polyfluoroaryl moiety.32,33 Slow reductive elimination prevents the formation of the α-substituted product and enables its return to the active species. When C–H bond cleavage occurs at the β-position, reductive elimination is relatively fast and yields the β-substituted product. Finally, the Ag salt re-oxidises the Pd(0) to Pd(II). Therefore, we hypothesised that the unique regioselectivity of the reaction is determined by the reductive elimination step34 rather than C–H bond cleavage. To obtain evidence supporting this hypothesis, model α- and β-biaryl Pd complexes were synthesised and compared in terms of reactivity in reductive elimination reactions (Scheme 3). Both biaryl Pd complexes with a N,N,N,N-tetramethylethylenediamine (TMEDA) supporting ligand were successfully prepared as model complexes (Scheme S1, ESI†).32 These model complexes were heated at 120 °C in CPME, and the reaction progress was monitored for 30 min. The α-biaryl model complex remained unreacted after 30 min (Scheme 3 and Fig. S3 and S4, ESI†). The stability of the α-biaryl model complex is considerably higher than that of similar biaryl complexes in the literature.32 In contrast, the reaction of the β-biaryl model complex resulted in a 92% yield of the reductive elimination product, a decomposed product (naphthalene, 7%), and no residual complex after 10 min (Fig. S5 and S6, ESI†). These results show a large difference in the reductive elimination rates of the α- and β-biaryl Pd complexes and support the proposed mechanism.
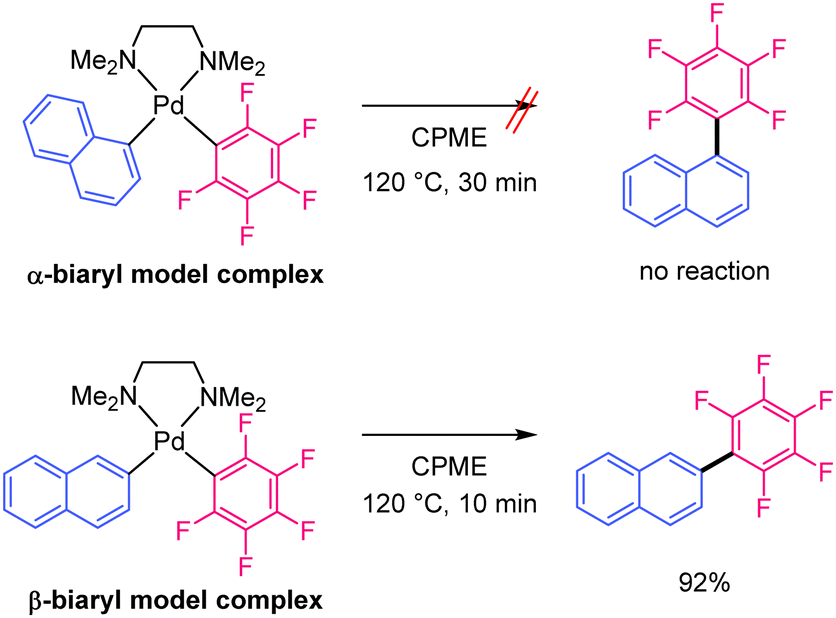 | ||
| Scheme 3 Reductive elimination reaction of the α- and β-biaryl model Pd complexes. | ||
In this study, Pd-catalysed CDC reactions of simple aromatic molecules with polyfluoroarenes were developed. We successfully introduced multiple polyfluoroarenes into naphthalene and anthracene at sterically favourable positions (β-position of naphthalene). An investigation of the reaction mechanism revealed that the unique regioselectivity was determined by the reductive elimination step rather than the C–H bond cleavage step. As this unique regioselectivity complements previous methodologies, we plan to apply this reaction to synthesise functional PAHs and semiconducting materials in future.
The authors thank the Chemical Analysis Center of the University of Tsukuba for the measurements of NMR, the single-crystal X-ray diffraction, and the elemental analysis. This work was partly supported by SEI GROUP CSR Foundation, JST A-STEP Grant Number JPMJTM20BT, and JST SPRING, Grant Number JPMJSP2124.
Conflicts of interest
There are no conflicts to declare.References
- Lecture Notes in Chemistry, ed. Y. Nishihara, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, vol. 80 Search PubMed.
- H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147–1164 CrossRef CAS.
- L. C. Campeau, D. R. Stuart and K. Fagnou, Aldrichimica Acta, 2007, 40, 35–41 CAS.
- Y. F. Zhang and Z. J. Shi, Acc. Chem. Res., 2019, 52, 161–169 CrossRef CAS PubMed.
- Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66–81 CrossRef CAS PubMed.
- P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K. S. Yeung and J. Q. Yu, Nature, 2017, 551, 489–493 CrossRef CAS PubMed.
- D. R. Stuart and K. Fagnou, Science, 2007, 316, 1172–1176 CrossRef CAS PubMed.
- K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 11904–11905 CrossRef CAS PubMed.
- C. Y. He, S. Fan and X. Zhang, J. Am. Chem. Soc., 2010, 132, 12850–12852 CrossRef CAS PubMed.
- A. Facchetti, M. H. Yoon, C. L. Stern, H. E. Katz and T. J. Marks, Angew. Chem., Int. Ed., 2003, 42, 3900–3903 CrossRef CAS PubMed.
- S. Ando, J. I. Nishida, E. Fujiwara, H. Tada, Y. Inoue, S. Tokito and Y. Yamashita, Chem. Mater., 2005, 17, 1261–1264 CrossRef CAS.
- Z. Qin, H. Gao, J. Liu, K. Zhou, J. Li, Y. Dang, L. Huang, H. Deng, X. Zhang, H. Dong and W. Hu, Adv. Mater., 2019, 31, 1903175 CrossRef PubMed.
- J. Liu, H. Zhang, H. Dong, L. Meng, L. Jiang, L. Jiang, Y. Wang, J. Yu, Y. Sun and W. Hu, Nat. Commun., 2015, 6, 10032 CrossRef CAS PubMed.
- J. Liu, H. Dong, Z. Wang, D. Ji, C. Cheng, H. Geng, H. Zhang, Y. Zhen, L. Jiang, H. Fu, Z. Bo, W. Chen, Z. Shuai and W. Hu, Chem. Commun., 2015, 51, 11777–11779 RSC.
- Y. Wei and W. Su, J. Am. Chem. Soc., 2010, 132, 16377–16379 CrossRef CAS PubMed.
- H. Li, J. Liu, C. L. Sun, B. J. Li and Z. J. Shi, Org. Lett., 2011, 13, 276–279 CrossRef CAS PubMed.
- M. D. Lotz, N. M. Camasso, A. J. Canty and M. S. Sanford, Organometallics, 2017, 36, 165–171 CrossRef CAS.
- Y. Shimoyama, J. Kuwabara and T. Kanbara, ACS Catal., 2020, 10, 3390–3397 CrossRef CAS.
- T. Bhattacharya, S. Dutta and D. Maiti, ACS Catal., 2021, 11, 9702–9714 CrossRef CAS.
- C. Y. He, Q. Q. Min and X. Zhang, Organometallics, 2012, 31, 1335–1340 CrossRef CAS.
- L. Xing, J.-R. Liu, X. Hong, K. N. Houk and C. K. Luscombe, J. Am. Chem. Soc., 2022, 144, 2311–2322 CrossRef CAS PubMed.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef.
- R. Li, L. Jiang and W. Lu, Organometallics, 2006, 25, 5973–5975 CrossRef CAS.
- F. Jafarpour, M. Ayoubi-Chianeh, M. Abbasnia and M. Azizzade, Catal. Sci. Technol., 2017, 7, 2930–2934 RSC.
- A. M. Wagner, A. J. Hickman and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 15710–15713 CrossRef CAS PubMed.
- M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS PubMed.
- K. Yamazaki, J. Kuwabara and T. Kanbara, Macromol. Rapid Commun., 2013, 34, 69–73 CrossRef CAS PubMed.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Org. Chem., 2012, 77, 658–668 CrossRef CAS PubMed.
- D. Kim, G. Choi, W. Kim, D. Kim, Y. K. Kang and S. H. Hong, Chem. Sci., 2021, 12, 363–373 RSC.
- A. Petit, J. Flygare, A. T. Miller, G. Winkel and D. H. Ess, Org. Lett., 2012, 14, 3680–3683 CrossRef CAS PubMed.
- K. Osakada, H. Onodera and Y. Nishihara, Organometallics, 2005, 24, 190–192 CrossRef CAS.
- V. Salamanca and A. C. Albéniz, Org. Chem. Front., 2021, 8, 1941–1951 RSC.
- T. Ohmura, K. Oshima, H. Taniguchi and M. Suginome, J. Am. Chem. Soc., 2010, 132, 12194–12196 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2099539 and 2114882. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04655e |
| This journal is © The Royal Society of Chemistry 2022 |