
Stepwise structure evolution from 2D cluster-based frameworks to silver(I) clusters†
Qiu-Xu
Zang
,
Zhao-Yang
Wang
,
Yao
Li
,
Xi-Ming
Luo
 ,
Hai-Yang
Li
,
Ya-Nan
Si
* and
Shuang-Quan
Zang
*
,
Hai-Yang
Li
,
Ya-Nan
Si
* and
Shuang-Quan
Zang
*
Henan Key Laboratory of Crystalline Molecular Functional Materials, Henan International Joint Laboratory of Tumor Theranostical Cluster Materials, Green Catalysis Center, and College of Chemistry, Zhengzhou University, Zhengzhou 450001, China
First published on 29th September 2022
Abstract
A pair of 2D chiral frameworks, R-Ag5-1 and S-Ag5-1, with orange fluorescence were successfully synthesized. They were transformed to a thermodynamically favored denser stacking, R-Ag5-3 and S-Ag5-3, accompanied by a change in the luminescence and activating the CPL signal. The capture and structural determination of the intermediates (R-Ag5-2 and S-Ag5-2) reveal that the coordination instability of Na+ ions is the main cause of this transition.
Owing to their atomically precise structures and unique physicochemical properties, metal clusters have a wide range of potential applications in luminescence, catalysis, biomedicine, chirality, and sensing.1,2 Besides, metal clusters have been noticed to bridge molecular and supramolecular chemistry, where different types of inter-cluster interactions, such as chemical bonding (Au–Ag, Ag–Ag, Ag–S), weak interactions (π⋯π interactions, electrostatic, hydrogen bonding, van der Waals interactions), and organic bridging ligands (e.g., 4,4′-bipyridyl), have an effect on the self-assembly mode of the cluster building blocks and their performance.3–12
In the recent past, the self-assembly of metal clusters induced by cations has received special attention due to their abundant assembly modes. Not only can cations act as counterions of electronegative clusters to control the synthesis of metal clusters, they can also induce the assembly of metal clusters to cluster-based supracrystals.13–16 For example, the shape of Ag44(p-MBA)304− changed from rhombohedral to octahedral by changing the p-MBA ligand counterions from H+ to Cs+.13 In addition, Zhu et al. assembled Ag29(SSR)12(PPh3)4 nanoclusters through Cs+ cations into distinct arrays (1D linear chains, 2D grid networks, and 3D superstructures), which not only changed the crystal-line packing modes but also manifested distinguishable optical properties.14
Chirality is omnipresent in nature and extends into much of modern science.17,18 Because of their abundant charge-transfer pathways and tunable chiral excited-state structures, metal clusters are excellent candidates for chiral materials.6 In general, the chiral transfer from ligands to metal cores is an efficient strategy for generating chiral metal clusters.19 Although significant research effort on chiral materials has been made, the chiral evolution from molecules to nanostructures, as well as the corresponding influence on their properties, has not yet been clearly illustrated.20 Therefore, the establishment of chiral models with atomically precise structures is crucial to the development of the chiral field.
Herein, we report the effect of Na+ ions on the formation and structural transformation of metal cluster-based assemblies, and explore the atomic-scale structure–property correlation. First, a pair of two-dimensional (2D) chiral frameworks [NaAg5(R/S-L)6(H2O)2(CH3OH)]·Na[Ag5(R/S-L)6] (denoted as R-Ag5-1, S-Ag5-1) built with R/S-tert-butyl-3-mercaptopyrrolidine-1-carboxylate (R-L, S-L)-protected Ag5 clusters bridged by Na+ were prepared. Notably, in the channels of the frameworks were orderly arranged discrete anion (Ag5L6)− clusters (Ag5 clusters for short). Interestingly, the orange fluorescent Ag5-1 transformed to Na[Ag5(R/S-L)6] (denoted as R-Ag5-3, S-Ag5-3) with bright green fluorescence under ambient conditions. Structural analysis of the intermediate Na[Ag5(R/S-L)6] (denoted as R-Ag5-2, S-Ag5-2) reveals that the coordinated methanol and water molecules of the sodium nodes gradually leave upon evaporation of the solvent molecules, resulting in disordering of the Na+ ions and then to a thermodynamically stable dense packing pattern. Migration of the Na+ cations in the framework leads to a change in the inter-cluster interactions, which not only induces a new CD signal but also successfully activates the CPL signal.
Colorless hexagonal crystal of Ag5-1 crystallizes in the chiral trigonal space group P3 with Flack parameters of 0.011(3) (R-Ag5-1) and 0.009(4) (S-Ag5-1) at 200 K, indicating homochiral molecular stacking. Take the structure of S-Ag5-1, for instance. The chiral Ag5 clusters are linked by Na+ ions through coordination bonds to afford a 2D structure in which discrete Ag5 clusters are arrayed in line in the channels (Fig. 1b). The layers further adopt an AA stacking mode and form 3D supramolecular structures with channels along the c-axis (Fig. 1c). The distance between two adjacent layers (from kernel Ag to kernel Ag) is ca. 9.125 Å (Fig. S5, ESI†)
 | ||
| Fig. 1 Structure of Ag5-1. (a) Ag5 cluster-based units of R-Ag5-1 (left) and S-Ag5-1 (right) in the host frameworks with mirror symmetry; (b) 2D laminar structure of S-Ag5-1 with ordered chiral Ag5 clusters in the channels viewed along the c-axis; (c) stacked schematic illustration of the 2D laminar structure of S-Ag5-1 viewed along the c-axis; (d) adjacent layers are stacked together through hydrogen-bond interactions viewed from b-axis; and (e) hydrogen bonds around a discrete Ag5 cluster in the channel viewed along the c-axis. Color labels: green and orange, Ag; yellow, S; blue, N; red, O; gray, C; white, H; azure, Na. The bright green dotted lines represent hydrogen bonds. | ||
The node of the metal clusters composed of five Ag atoms is linked through Ag(I)⋯Ag(I) interactions and is shaped in a triangular prism with the six S atoms, where the S atoms are located at the vertices, two Ag atoms at the center of the triangular planes and three Ag atoms at the center of the three side edges (Fig. 1a). The thiol ligands are spirally arranged on the top and bottom of the triangular prism Ag5S6 in the opposite direction, which is similar to a trilobal fan (Fig. S6, ESI†). The Ag–Ag distances of the Ag5 clusters in the host framework range from 3.205 to 3.366 Å with an average of 3.287 Å, which is less than twice the van der Waals radius of the Ag atom (2 × 1.72 Å), indicating the existence of argentophilic interactions.8 In addition, the difference in the six Ag–Ag bond lengths leads to the formation of a twisted triangular biconical structure for the five silver atoms of the core. The S atoms of all six ligands uniformly adopt the μ2-η1,η1 coordination mode connecting two Ag atoms. The Ag–S bonds range from 2.360 to 2.490 Å with an average bond length of 2.421 Å. By contrast, the chiral Ag5 clusters in the channels form a relatively perfect triangular biconical structure wherein the lengths of Ag–Ag bonds are 3.287 Å and 3.311 Å (Fig. S7, ESI†).
Each Na+ is five-coordinated by two carbonyl groups from adjacent Ag5 cluster-based nodes, two water molecules and one methanol molecule (Fig. S8, ESI†). From the c-axis, carbonyl groups on two of the three ligands in the upper layer of each triangular prism Ag5S6 node coordinate with Na+, and only one carbonyl group in the lower layer participates in the coordination (Fig. S9, ESI†). Three carbonyl groups of every Ag5S6 node in the framework and six carbonyl groups of every Ag5 cluster in the channels are unutilized according to the coordination requirement of Na+, thus some of them form O⋯H–O hydrogen bonds with coordinate water molecules. As shown in Fig. 1d, adjacent layers are stacked together through O⋯H–O (2.001–2.147 Å) interactions viewed from the b-axis. In addition, the stable presence of guest Ag5 clusters in the channels is also due to weak hydrogen-bonding interactions, where the length of the O⋯H–O hydrogen bonds is 1.909 Å (Fig. 1e).
It is of note that the PXRD pattern of the dried sample stored for several hours under ambient conditions showed considerable changes; meanwhile, the emission color varied from weak orange to intense green. Since the dried crystals (hereafter labelled R-Ag5-3 and S-Ag5-3) broke up completely and SCXRD analysis was not possible, we tried to cultivate the final-state single-crystal structure to figure out what the transition was. Fortunately, replacing sodium borohydride with hydrazine hydrate resulted in single crystals (hereafter labelled R-Ag5-3′ and S-Ag5-3′) with good quality and the same green emission as the cracked Ag5-3 crystals. Further electrospray ionization mass spectrometry characterization and PXRD patterns showed the structural consistency of S-Ag5-3 and S-Ag5-3′ in solution and the solid state, respectively. As shown in Fig. S11 (ESI†), the dominant peaks in dilute solutions of S-Ag5-3 and S-Ag5-3′ corresponded to [Ag5(C9H16NO2S)6]− with m/z 1753.07 (calculated m/z 1753.06). Moreover, the PXRD patterns showed that the characteristic peaks of S-Ag5-3 and S-Ag5-3′ are identical, suggesting the uniform structure of the two (Fig. 2a). Ag5-3′ crystallizes in the triclinic chiral space group P1 with Flack parameters of 0.023(12) (R-Ag5-3′) and 0.003(17) (S-Ag5-3′) (Table S5, ESI†). The Ag5 cluster-based units in S-Ag5-3′ are more tightly packed in a staggered stacking pattern than that in S-Ag5-1 (Fig. S12, ESI†). In addition, compared with S-Ag5-1, the triangular prism of the Ag5 cluster-based units in S-Ag5-3′ is distorted and the separation between the two Ag atoms at the center of the top and bottom planes decreased from 4.651 Å to 4.427 Å, where all the Ag–Ag and Ag–S bond lengths are different. Fig. S13 (ESI†) depicts the metal cores of the two enantiomers, displaying near-perfect mirror images of R-Ag5-3′ and S-Ag5-3′. As shown in Fig. 2b, temperature-dependent PXRD analysis corroborates the high thermostability of S-Ag5-3 up to 150 °C in air, which is consistent with the thermogravimetric analysis (TGA) curves (Fig. S14, ESI†). This is a rare property in sliver clusters protected by mercaptan ligands, including normal-valence and mixed-valence clusters.19
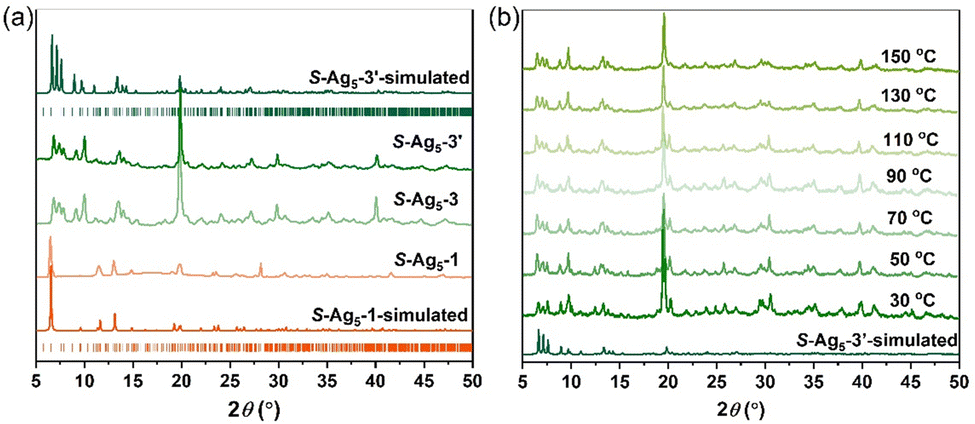 | ||
| Fig. 2 (a) PXRD patterns of S-Ag5-1, S-Ag5-3 and S-Ag5-3′; and (b) temperature-dependent PXRD patterns of S-Ag5-3 tested in air. | ||
The compositions of Ag5-1 and Ag5-3 are similar, but the packing patterns of the Ag5 cluster-based units have changed dramatically. To understand this transition, we succeeded in obtaining the crystal structures of the intermediates (R-Ag5-2 and S-Ag5-2) with excellent quality by soaking the Ag5-1 crystal in anhydrous methanol for one week, and whose crystal parameters were consistent with those measured via an in situ heating method (see ESI† for details). Ag5-2 shows orange fluorescence consistent with Ag5-1 at room temperature but bright green fluorescence at low temperature. Therefore, the structures of R-Ag5-2 and S-Ag5-2 were further determined via SCXRD at 200 K and 295 K to explore the relationship between the fluorescence and the structure. The results indicate that Ag5-2 crystallizes in the same trigonal chiral space group P3 as Ag5-1 at both 200 K and 295 K (Tables S3 and S4, ESI†). Ag5-2 displays a similar lattice structure to Ag5-1 but where Na+ ions are disordered in the structure and only serve as counterions (Fig. 3). In comparison to that in Ag5-1, all the Ag5 cluster-based units in the structure of Ag5-2 are identical. For example, the interlayer spacing (from kernel Ag to kernel Ag) of S-Ag5-2 is expanded to 9.297 Å (Fig. S15 and S16, ESI†). A comparison of the structures of S-Ag5-2 at 200 K and 295 K demonstrates that considerable bond contractions in each triangular prism Ag5S6 node were observed, with the Ag–Ag bond distance shortened by 0.014 Å from 3.306 Å (200 K) to 3.292 Å (295 K), as well as shrinkage of the Ag–S bond lengths at the triangular planes and three side edges (Fig. S17, ESI†). We speculate that the changing length of the Ag–Ag and Ag–S bonds is the main cause of the luminescence change. The capture and structural determination of the intermediate Ag5-2 revealed that the loss of coordinated solvent molecules leads to dissociation of the 2D framework and subsequently to denser and more thermodynamically stable stacking.
 | ||
| Fig. 3 Schematic of the transformation from S-Ag5-1 to S-Ag5-2, and then to S-Ag5-3 viewed along the c-axis. Color labels: green and orange, Ag; yellow, S; blue, N; red, O; gray, C; white, H; azure, Na. Some hydrogen atoms are omitted for clarity. | ||
The photophysical properties of the Ag5-1 and Ag5-3 were fully characterized via their UV-vis absorption and photoluminescence spectra. Solid-state Ag5-1 and Ag5-3 exhibit similar UV-vis absorption features with a band edge at 400 nm, where the peak below 300 nm can be assigned to the n → π* transitions of the ligands, and the diffuse reflection peak at 300–400 nm can be attributed to electronic transitions from the (Ag–S) σ orbital to the empty π* orbital (Fig. 4a and Fig. S18, ESI†). The Ag5-1 exhibit faint orange emission centered at 600 nm under excitation at 361 nm, while the emission of the Ag5-3 is blue-shifted to 533 nm with excitation at 350 nm (Fig. 4b), and the quantum yield is 0.54%. The lifetimes of R-Ag5-3 and S-Ag5-3 are 3.2 μs and 3.3 μs, respectively, which indicate that the emission originates from spin-forbidden triplet-state phosphorescence (Fig. S19, ESI†).21
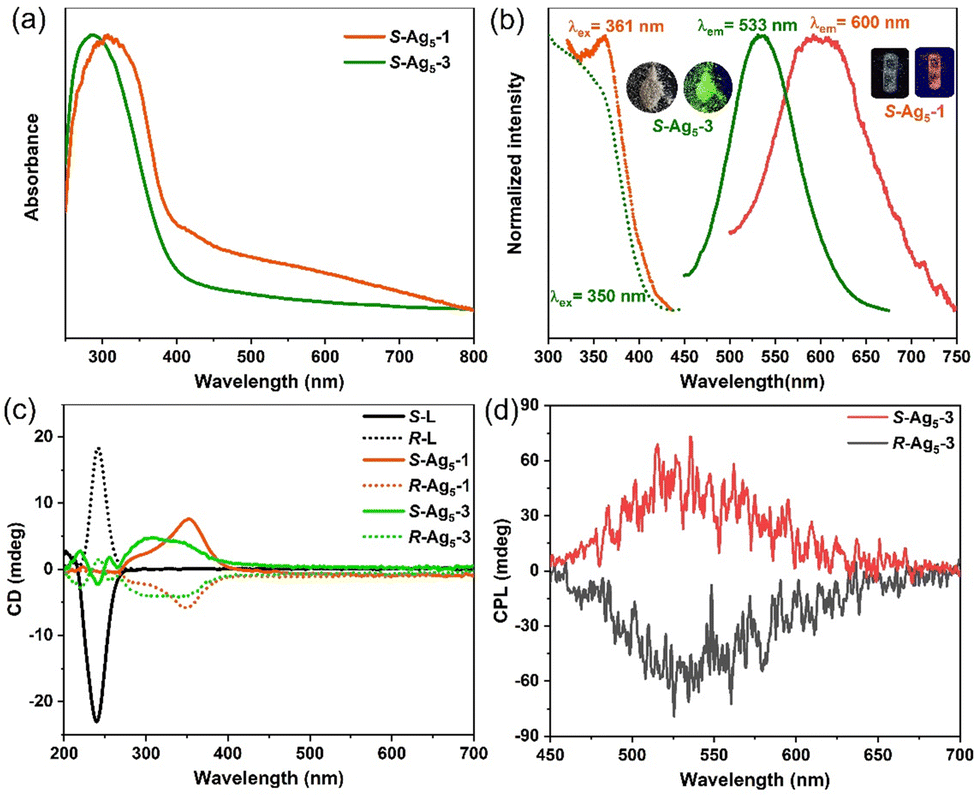 | ||
| Fig. 4 (a) UV-vis absorption spectra of solid-state S-Ag5-1 and S-Ag5-3 at RT. (b) Normalized excitation and emission spectra of solid-state S-Ag5-1 (orange) and S-Ag5-3 (green) at RT. (c) CD spectra of enantiomers R/S-Ag5-1, R/S-Ag5-3 and R/S-L in the solid state. (d) CPL spectra of solid-state enantiomers R/S-Ag5-3. | ||
To further understand the complex photophysical properties, density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations based on the single-crystal structure of R-Ag5-3 were performed, where the highest occupied molecular orbital (HOMO) is mainly located on the S atoms of the peripheral ligands with partial distribution on the core Ag atoms, while the lowest unoccupied molecular orbitals (LUMOs) are mainly located on the Ag5 core (Fig. S20, ESI†). The calculated UV-vis absorption spectrum of R-Ag5-3 agreed well with the experimental spectrum, demonstrating the consistency of structure in solution (Fig. S21, ESI†). The absorption band at 210 nm mainly belongs to the HOMO−16 → LUMO+1 and HOMO−3 → LUMO+1 transitions, proving that the electronic transitions mainly involve the Ag atoms and the six S atoms of the ligands.
Given the chiral structures and luminescence of the R/S-Ag5-1 enantiomers and R/S-Ag5-3 enantiomers, the circular dichroism (CD) and circularly polarized luminescence (CPL) were investigated. The CD spectra of the R/S-Ag5-1 enantiomers and R/S-Ag5-3 enantiomers both exhibit a strong Cotton effect in the range of 219–450 nm (Fig. 4c). Specifically, the symmetric CD signals of the R/S-Ag5-1 enantiomers appear at 286 and 352 nm. Enantiomers R/S-Ag5-3 show a series of bands at 219, 240, 255, 304, and 340 nm, where the CD signal at 240 nm belongs to the chiral ligands, while others are attributed to the chiral cluster structure induced by the asymmetric arrangement of the chiral ligands (Fig. 4c). The strong symmetric CPL response of the solid-state R/S-Ag5-3 enantiomers, in striking contrast to non-CPL-active R/S-ligands, appears in the same wavelength region as their emission band centered at 533 nm, which indicates that emitted light from the lowest excited states in R/S-Ag5-3 is polarized in opposite directions (Fig. 4d). The maximum glum values of the R/S-Ag5-3 enantiomers were measured to be approximately ±5 × 10−3 (Fig. S22, ESI†), which are comparable to those of reported CPL-based gold clusters.22 Overall, the consistency between the CD and UV-vis spectra, and between the CPL and PL spectra, together with the definite crystallographic structure enables us to understand the intrinsic optical activity of the chiral silver clusters at the atomic level.
In conclusion, the coordination of sodium ions with carbonyl groups results in the assembly of Ag5 clusters into a 2D framework structure. The framework Ag5-1 could be converted to intermediate Ag5-2 with disordered Na+ in the structure. Subsequently, Ag5-2 transformed to the thermodynamically favored more closely stacked product Ag5-3, accompanied by an orange-to-green emission conversion because of the distortion of the Ag5(R/S-L)6− kernel. In addition, R/S-Ag5-3 enantiomers showed unusual thermal stability and excellent CPL activity, as well as different CD signals from the R/S-Ag5-1 enantiomers. This work structurally elucidates the transformation process of Ag5 cluster-based assemblies, and provides new insights into the atom-precise interactions between the sodium ions and metal clusters.
This work was supported by the National Natural Science Foundation of China (No. 22105176, 92061201, 21825106, 21801228) and Zhengzhou University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Li, F. Song, D. Zhu, Y. Song, C. Zhou, F. Ke, L. Lu, X. Kang and M. Zhu, J. Am. Chem. Soc., 2022, 144, 4845–4852 CrossRef CAS PubMed.
- G. Li, X. Sui, X. Cai, W. Hu, X. Liu, M. Chen and Y. Zhu, Angew. Chem., Int. Ed., 2021, 60, 10573–10576 CrossRef CAS PubMed.
- P. Chakraborty, A. Nag, A. Chakraborty and T. Pradeep, Acc. Chem. Res., 2019, 52, 2–11 CrossRef CAS.
- H. Wu, X. He, B. Yang, C. C. Li and L. Zhao, Angew. Chem., Int. Ed., 2021, 60, 1535–1539 CrossRef CAS PubMed.
- Y. Jin, C. Zhang, X. Y. Dong, S. Q. Zang and T. C. W. Mak, Chem. Soc. Rev., 2021, 50, 2297–2319 RSC.
- G. Deng, B. K. Teo and N. Zheng, J. Am. Chem. Soc., 2021, 143, 10214–10220 CrossRef CAS PubMed.
- Q. Yao, Z. Wu, Z. Liu, Y. Lin, X. Yuan and J. Xie, Chem. Sci., 2020, 12, 99–127 RSC.
- A. Ebina, S. Hossain, H. Horihata, S. Ozaki, S. Kato, T. Kawawaki and Y. Negishi, Nanomaterials, 2020, 10, 1105 CrossRef CAS.
- S. Chen, W. Du, C. Qin, D. Liu, L. Tang, Y. Liu, S. Wang and M. Zhu, Angew. Chem., Int. Ed., 2020, 59, 7542–7547 CrossRef CAS PubMed.
- X. Wei, X. Kang, Q. Yuan, C. Qin, S. Jin, S. Wang and M. Zhu, Chem. Mater., 2019, 31, 4945–4952 CrossRef CAS.
- Z. Lei, X. L. Pei, Z. G. Jiang and Q. M. Wang, Angew. Chem., Int. Ed., 2014, 53, 12771–12775 CrossRef CAS PubMed.
- A. Nag, P. Chakraborty, M. Bodiuzzaman, T. Ahuja, S. Antharjanam and T. Pradeep, Nanoscale, 2018, 10, 9851–9855 RSC.
- Q. Yao, Y. Yu, X. Yuan, Y. Yu, D. Zhao, J. Xie and J. Y. Lee, Angew. Chem., Int. Ed., 2015, 54, 184–189 CrossRef.
- X. Wei, X. Kang, Z. Zuo, F. Song, S. Wang and M. Zhu, Natl. Sci. Rev., 2021, 8, nwaa077 CrossRef.
- A. Som, I. Chakraborty, T. A. Maark, S. Bhat and T. Pradeep, Adv. Mater., 2016, 28, 2827–2833 CrossRef.
- Y. Jin, S. Li, Z. Han, B. J. Yan, H. Y. Li, X. Y. Dong and S. Q. Zang, Angew. Chem., Int. Ed., 2019, 58, 12143–12148 CrossRef.
- C. Zeng, Y. Chen, K. Kirschbaum, K. J. Lambright and R. Jin, Science, 2016, 354, 1580–1584 CrossRef PubMed.
- H. Shen, Z. Xu, L. Wang, Y. Z. Han, X. Liu, S. Malola, B. K. Teo, H. Hakkinen and N. Zheng, Angew. Chem., Int. Ed., 2021, 60, 22411–22416 CrossRef.
- Z. Han, X. Y. Dong, P. Luo, S. Li, Z. Y. Wang, S. Q. Zang and T. C. W. Mak, Sci. Adv., 2020, 6, eaay0107 CrossRef PubMed.
- W. Ma, L. Xu, A. F. de Moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 117, 8041–8093 CrossRef PubMed.
- G. Li, Z. Lei and Q.-M. Wang, J. Am. Chem. Soc., 2010, 132, 17678–17679 CrossRef PubMed.
- L. Shi, L. Zhu, J. Guo, L. Zhang, Y. Shi, Y. Zhang, K. Hou, Y. Zheng, Y. Zhu, J. Lv, S. Liu and Z. Tang, Angew. Chem., Int. Ed., 2017, 56, 15397–15401 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2192030–2192037. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04623g |
| This journal is © The Royal Society of Chemistry 2022 |