
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical exchange of labile protons by deuterium enables selective detection of pharmaceuticals in solid formulations†
Claire
Welton‡
a,
Parth
Raval‡
a,
Julien
Trébosc
b and
G. N. Manjunatha
Reddy
 *a
*a
aUniversity of Lille, CNRS, Centrale Lille Institut, Univ. Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-59000, Lille, France. E-mail: gnm.reddy@univ-lille.fr
bUniversity of Lille, CNRS, INRAE, Centrale Lille, Univ. Artois, FR 2638 – IMEC – Institut Michel-Eugène Chevreul, F-59000, Lille, France
First published on 14th September 2022
Abstract
Chemically assisted swapping of labile protons by deuterons is presented for amino acids, polysaccharides, pharmaceutical compounds, and their solid formulations. Solid-state packing interactions in these compounds are elucidated by 1H–2H isotope correlation NMR spectroscopy (iCOSY). A minuscule concentration of dopamine, 5 wt% or ∼100 μg, in a solid formulation can be detected by 2H NMR at 28.2 T (1H, 1200 MHz) in under a minute.
Site-selective deuteration of small molecules has emerged as an indispensable tool in a variety of research areas such as drug development, structural biology, kinetic isotopic effects, and probing biochemical/enzymatic reactions.1–6 In particular, deuterated active pharmaceutical ingredients (d-APIs or deuterium isotopologues) have received significant attention owing to their unique physicochemical properties. The d-APIs tend to significantly alter pharmacokinetic properties by retarding the metabolism in vivo, which can extend the lifetime of therapeutics, enabling lower dosing than the conventional APIs to achieve the same physiological activity, selectivity and biochemical potency.3–6 In addition, deuterated amino acids and nucleotides have garnered interest in chemical biology for probing enzymatic/metabolic pathways.7,8
Deuterium (2H) isotopic labelling can be achieved by de-novo synthesis of small molecules using heterogeneous catalysis or enzymatic reactions.9–12 Nanostructured iron catalysts have been employed to selectively deuterate CH sites in (hetero)arenes and heterocycles such as anilines, phenols, and indoles.9,13 Dual-protein catalysis has been shown to deuterate Cα and Cβ sites in amino acids.14 Electrochemical methods,15 and photocatalysis have also been used as a green chemistry alternative for the deuteration of organic compounds.16 Isotopic 2H enrichment of labile sites such as NHn (n = 1–3) and OHn (n = 1, 2) groups can be achieved by treating the samples with deuterated solvents, facilitating the NMR analysis of biomacromolecules,17 though it leads to low yields of deuteration. Here, we present a facile chemical exchange protocol to deuterate labile protons in pharmaceutical compounds by dissolution and crystallization from a 2H-enriched solvent at different temperatures, yielding up to 90% deuterium substitution. The high degree of H/D exchange enabled the structural elucidation of drug molecules in solid formulations by magic-angle spinning (MAS) NMR spectroscopy.
The multistep approach we propose for site-specific deuteration in small molecules is presented in Fig. 1. The process begins with the dissolution of small molecules in deuterated solvents, in which the labile protons in amine, imide, and hydroxyl groups are exchanged by deuterium, but not the CHn (n = 1–3) sites. The rate of H/D exchange depends on proton lability and solubility, as well as the crystallization temperature. The extent of H/D exchange can be assessed by analysing 1H MAS NMR spectra of the powder before and after deuteration (ESI,† Section 2). For L-histidine·HCl·H2O and dopamine·HCl, degree of deuteration (%) is presented in Fig. 1c (ESI,† Fig. S1) for different sites as a function of temperature. Crystallization from D2O at 295 (±3) K enables H/D exchange yields of 19% and 17% for the NH3 sites in L-histidine·HCl·H2O, and dopamine·HCl, respectively. Crystallization at 373 (±3) K yields a more facile exchange, resulting in 92% and 77% of deuteration for the same sites. A high degree of deuteration can be achieved in a single cycle where the temperature is not a concern. However, multiple cycles of deuteration and recrystallization may be required for the biological systems at relatively low temperatures (<290 K). In contrast, cellulose is poorly soluble in water and exhibits low affinity towards H/D exchange (Fig. 1c, and ESI† Fig. S2) even at high temperatures. It is also expected to be the case for saturated fatty acids, polymer micelles, and glidants used in pharmaceutical formulations. Therefore, the presented approach can be used to isotopically enrich labile hydrogen atoms in the APIs without significantly deuterating excipients. Below, we illustrate how this method can be used to selectively detect histidine and dopamine in solid formulations.
 | ||
| Fig. 1 (a) A schematic of site-selective H–D exchange for small molecules, (b) molecular structures of L-histidine·HCl·H2O, dopamine·HCl and cellulose, and (c) the degree of deuteration measured by 1H MAS NMR spectroscopy. Grey and green bars correspond to the deuteration (%) obtained at 295 K and 373 K. *contribution from OH. | ||
Although deuterated compounds have been widely investigated in the context of medicinal chemistry, 4 the molecular-level understanding of amorphous solid dispersions has been exceedingly challenging to obtain due to the compositional and structural heterogeneities associated with APIs and excipients. Solid-state NMR spectroscopy has been used to elucidate local structures, packing interactions, and (pseudo)polymorphism in pharmaceutical compounds.18,19 Specifically, 2D experiments such as 1H–X (X = 1H, 13C, 14/15N, 19F, and 35Cl) enabled the local structures and interactions in neat APIs and solid formulations to be elucidated.19–24 One notable example is the use of 2D 1H–14N and 14N-filtered 1H–1H correlation experiments coupled with spin-diffusion process for detecting APIs in solid formulations.21,23 The proposed 2D 1H–2H iCOSY experiment operates in a manner akin to what is observed in 1H–14N correlation experiments, nonetheless the H/D exchange of –NHn and –OHn manifests more 2H sites to be excited and resolved in APIs.
Fig. 2a compares 1H MAS spectra of neat and partially deuterated histidine, showing differences in the peak intensities for NH(5) and NH(7), NH3(1), and water (w) protons. In the 2H MAS NMR spectrum of the latter compound (Fig. 2b), strong intensity peaks corresponding to ND3 and water-d peaks are observed, and weak intensity broad peaks are appeared for D5 and D7 sites. For small molecules, sensitivity and resolution aspects of 2H MAS NMR have previously been discussed.25–28 Molecular motions reduce the quadrupolar interactions, allowing narrow 2H peaks to be detected.25,26,29,30 Resolution can be substantially improved by MAS in conjunction with high magnetic fields (ESI,† Fig. S3 and S4), enabling the isotropic 2H chemical shifts as well as quadrupolar couplings to be measured and compared for different sites (ESI,† Fig. S5 and Table S1).
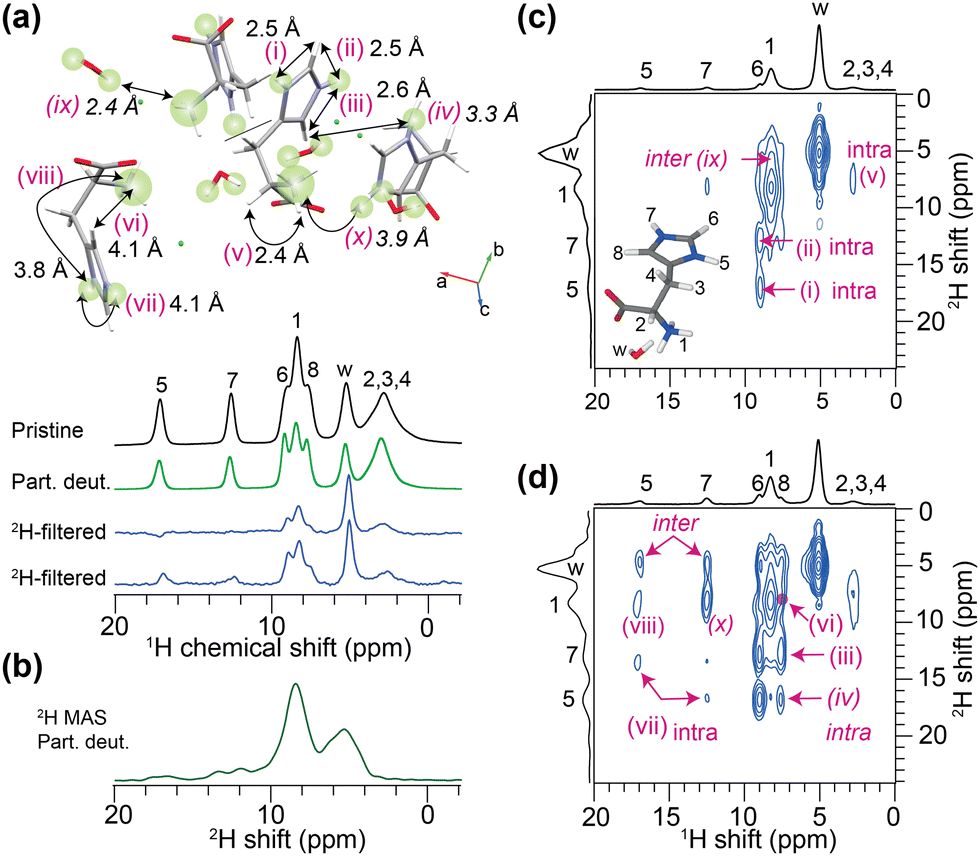 | ||
| Fig. 2 (a) 1D 1H MAS spectra of L-histidine·HCl·H2O: pristine (black) and deuterated (green) with a 2H-filter using different recoupling times (blue), along with (b) a 2H MAS spectrum of the same compound. 2D 1H–2H iCOSY spectra acquired with (c) short (133.3 μs) and (d) long (200 μs) recoupling times. 1H–2H distances are depicted in inset (a).§¶ All spectra were acquired at 18.8 T (1H = 800.1 MHz, 60 kHz MAS). | ||
A simple route to acquire 2D 1H–2H iCOSY spectra is to use a heteronuclear multiple-quantum coherence (HMQC) pulse sequence31,32 (ESI,† Fig. S1), in which the recoupling delay (τrcpl) can be adjusted to detect 2D peaks corresponding to 1H–2H sites separated by short (<3 Å) and mid-range (3–5 Å) distances. The blue spectra (Fig. 2a) illustrate that, a priori, by adjusting the 1H–2H recoupling time, all 1H peaks can be detected. By comparison, 2D iCOSY spectra acquired with 133.3 μs and 200 μs (Fig. 2c and d) recoupling times display peaks corresponding to short (<3 Å) and mid-range (>3 Å) 1H–2H proximities, as depicted in the inset (Fig. 2a).33 In Fig. 2c, 2D peaks associated with D5–H6, D7–H6/H8, ND3–CH2, CH2–D2O, and ND3–D2O are detected.§ In Fig. 2d, detection of the 2D peaks corresponding to through-space D5–H8 (iv) and ND3–H8 (vi) proximities with 1H–2H distances >4 Å is noteworthy.§ Meanwhile, one has to take into account that the 2H isotope substitution may lead to shorter bond distances.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ¶
¶
Next, we studied a solid formulation consisting of L-histidine·HCl·H2O and cellulose (20/80 wt%) using the 2D 1H–2H iCOSY approach. For this blend, 1D 1H (before and after deuteration), 1H{2H} and 2H MAS spectra are shown in the ESI† (Fig. S6), in which the 2H-filter suppresses the cellulose signals enabling the histidine peaks to be resolved. Fig. 3 presents 2D iCOSY spectra, whereby the spectrum acquired with a short recoupling time (100 μs, Fig. 3a) showed peaks associated with ND3/NH3 and H2O/D2O, whereas a long recoupling time (200 μs, Fig. 3b) allowed 2D peaks corresponding to the ND3-water, water-H5/H7/H8, water-CH2 and ND3–H5 proximities to be detected. In addition, weak intensity peaks associated with cellulose (cyan box) are observed. Key learning from this analysis is that the local structures of partially deuterated molecules in complex solid formulations can be resolved and identified.
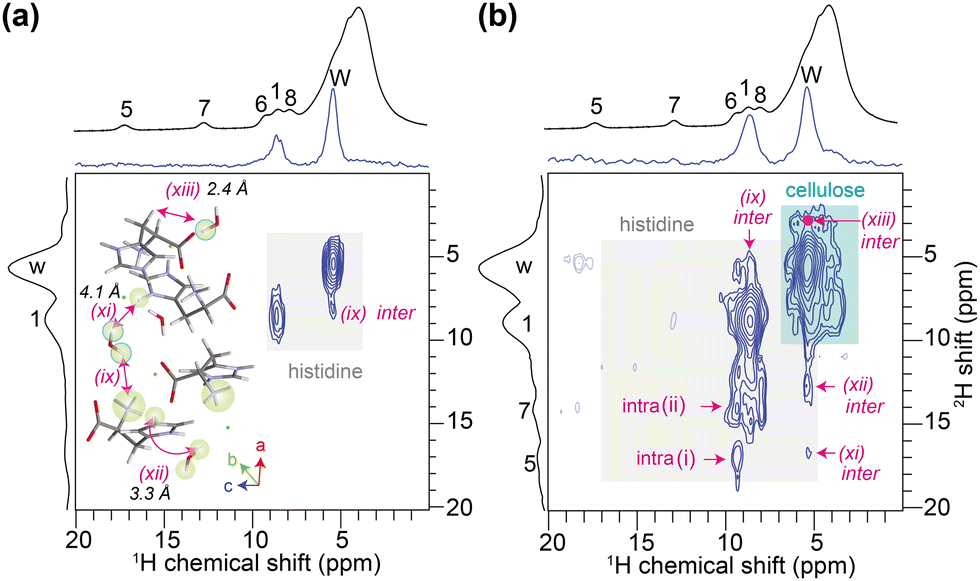 | ||
| Fig. 3 2D 1H–2H iCOSY spectra of partially deuterated histidine-cellulose (20/80 wt%) blend acquired with (a) 99.9 μs and (b) 199.8 μs of mixing time, plotted with the projections. 1D 1H MAS spectra (black) are presented in the top. | ||
The sensitivity and resolution capabilities of high-field 2H NMR for characterising drug formulations are further examined by analysing dopamine(DOPA)-cellulose blends, with different DOPA concentrations in the 5–30 wt% range. DOPA and its analogue levodopa (L-DOPA) have been widely investigated in the context of Parkinson's disease and related disorders.34Fig. 4 compares ssNMR spectra of DOPA and DOPA-cellulose blends, in which neat DOPA (Fig. 4a) displays a broad peak at ∼7.6 ppm due to an overlapped contribution from NH3 and aromatic protons, and partially resolved peaks at 2–4 ppm and 4–6 ppm are due to the CH2 (2, 2’, 3 and 3’) moieties. While all 1H peaks are observed in the 1H MAS spectra of pristine (black) and partially deuterated DOPA (green), the 1H{2H} MAS spectrum of the latter (blue) yields selective detection of the peak at ∼7.6 ppm (NH3/ND3 and OH/OD). Consequently, 2H MAS spectrum (green, bottom) displays a peak at ∼7.6 ppm, facilitating the analysis of chemical shifts and quadrupolar parameters of ND3 and OD (ESI,† Fig. S7, S8 and Table S2). For the same compound, the 2D 1H–2H iCOSY spectrum (Fig. 4b, and ESI† Fig. S9) shows a strong peak originating from NH3/ND3 and a weak intensity peak corresponding to ND3/CH2 proximities.33 In comparison, DOPA-cellulose blends exhibit severely overlapped 1H MAS NMR spectra (Fig. 4c) with peaks originating from cellulose. Notably, detection of the 1H peak at 7.8 ppm for 5 wt% DOPA (∼100 μg, for which ∼75% NH3 sites are deuterated) illustrates the resolving power of high field ssNMR (28.2 T) for the analysis of a complex solid form. However, structurally disordered arrangements of the excipients give rise to chemical shift distributions (2–7 ppm) that cannot be averaged out even under fast MAS at high fields. Since excipients do not exhibit a tendency towards H/D exchange, their broad spectral features can be easily eliminated by acquiring 2H NMR spectra (Fig. 4c, bottom). High-resolution 2H NMR spectra of DOPA in the blend can be detected in under a minute (∼10 seconds with a signal/noise of ∼20). Likewise, we acquired 1D 1H and 1H{2H} MAS (Fig. 4d, blue) and 2D 1H–2H iCOSY spectra to analyse the local structure of DOPA in the blend. In the 2D iCOSY spectrum (Fig. 4d, bottom), peaks correlating 2H (∼7.6 ppm) and 1H (5.9 and 3.4 ppm) chemical shifts are due to intramolecular proximity between the NH3 and CH2 groups (distances are shown in ESI,† Fig. S10). By comparison, low intensity 2D peaks between the 2H (∼5.5 ppm) and signals 1H (5.5–3.0 ppm) are expected to arise from the cellulose, and the 2D peak between 2H (∼5.5 ppm) and 1H (∼7.8 ppm) is likely to indicate the through-space ND3–CH2 proximities (DOPA) as well as the DOPA-cellulose proximities.
 | ||
| Fig. 4 Structure elucidation of DOPA and DOPA-cellulose blends. (a) top: 1D 1H MAS spectra of pristine (black) and partially deuterated (green) DOPA and with a 2H-filter (blue) and 2H NMR spectrum (bottom) of the same sample acquired at 18.8 T (1H = 800.1 MHz, 60 kHz MAS). (b) A 2D 1H–2H iCOSY spectrum of DOPA acquired at 18.8 T with τRCPL = 199.8 μs together with the crystal structure depicting the interatomic distances. (c) 1D 1H MAS spectra (top) of DOPA-cellulose blends with different concentrations of DOPA as indicated, and 2H NMR spectra (bottom) of the same compounds acquired at 28.2 T (1H = 1200.5 MHz, 60 kHz MAS). (d) A 1D 1H MAS spectrum of pristine (black) and a 2H-filtered (blue) spectrum of the DOPA-cellulose blend along with a 2D iCOSY spectrum acquired with the same conditions as in (b). Distances corresponding to (I–VII) are shown in ESI,† Fig. S10. | ||
In this communication we present a facile route to deuterate the labile protons in pharmaceutical compounds, while excipients do not exhibit this trend due to the poor solubility in water. Stimulated by the 2H enrichment, this study presents a 2D 1H–2H iCOSY technique for detecting APIs in solid formulations. Compared to conventional 2D 1H–14N HMQC, the 1H–2H iCOSY approach benefits from: (i) relatively large 1H–2H dipolar couplings than the 1H–14N dipolar couplings despite the low natural abundance of 2H (0.015%), (ii) smaller quadrupolar interactions in 2H than 14N sites, and (ii) there are more 2H{1H} sites that can be labelled in APIs than the naturally occurring 14N{1H} sites, which is an added advantage for the structure elucidation. Cross-polarization (CP) and spin-diffusion techniques provide further sensitivity enhancements in 2D 1H–2H iCOSY spectra.33,35 Therefore, the iCOSY approach is likely to see a promising future for solid-state characterization.
We acknowledge the financial support from the EU-H2020 Grant 795091 and IR INFRANALYTICS FR2040. Dr Pawlak is thanked for helping with DFT calculations.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Kopf, F. Bourriquen, W. Li, H. Neumann, K. Junge and M. Beller, Chem. Rev., 2022, 122, 6634–6718 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758–1784 CrossRef CAS PubMed.
- B. Czeskis, C. S. Elmore, A. Haight, D. Hesk, B. D. Maxwell, S. A. Miller, T. Raglione, K. Schildknegt, J. F. Traverse and P. Wang, J. Labelled Compd. Radiopharm., 2019, 62, 690–694 CrossRef CAS.
- S. H. DeWitt and B. E. Maryanoff, Biochemistry, 2018, 57, 472–473 CrossRef CAS PubMed.
- T. G. Gant, J. Med. Chem., 2014, 57, 3595–3611 CrossRef CAS.
- T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed.
- R. Agarwal, M. D. Smith and J. C. Smith, J. Chem. Theory Comput., 2020, 16, 2529–2540 CrossRef CAS.
- S. Hanashima, Y. Ibata, H. Watanabe, T. Yasuda, H. Tsuchikawa and M. Murata, Org. Biomol. Chem., 2019, 17, 8601–8610 RSC.
- R. Pony Yu, D. Hesk, N. Rivera, I. Pelczer and P. J. Chirik, Nature, 2016, 529, 195–199 CrossRef CAS.
- G. Prakash, N. Paul, G. A. Oliver, D. B. Werz and D. Maiti, Chem. Soc. Rev., 2022, 51, 3123–3163 RSC.
- T. R. Puleo, A. J. Strong and J. S. Bandar, J. Am. Chem. Soc., 2019, 141, 1467–1472 CrossRef CAS PubMed.
- Z. P. Vang, A. Reyes, R. E. Sonstrom, M. S. Holdren, S. E. Sloane, I. Y. Alansari, J. L. Neill, B. H. Pate and J. R. Clark, J. Am. Chem. Soc., 2021, 143, 7707–7718 CrossRef CAS.
- W. Li, J. Rabeah, F. Bourriquen, D. Yang, C. Kreyenschulte, N. Rockstroh, H. Lund, S. Bartling, A. E. Surkus, K. Junge, A. Brückner, A. Lei and M. Beller, Nat. Chem., 2022, 14, 334–341 CrossRef CAS PubMed.
- T. J. Doyon and A. R. Buller, J. Am. Chem. Soc., 2022, 144, 7327–7336 CrossRef CAS PubMed.
- P. L. Norcott, Chem. Commun., 2022, 58, 2944–2953 RSC.
- P. Ranjan, S. Pillitteri, E. V. Van Der Eycken and U. K. Sharma, Green Chem., 2020, 22, 7725–7736 RSC.
- B. Reif, J. Magn. Reson., 2012, 216, 1–12 CrossRef CAS PubMed.
- A. J. Rossini, C. M. Widdifield, A. Zagdoun, M. Lelli, M. Schwarzwälder, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2014, 136, 2324–2334 CrossRef CAS.
- M. Li, W. Xu and Y. Su, TrAC, Trends Anal. Chem., 2021, 135, 116152 CrossRef CAS.
- A. S. Tatton, T. N. Pham, F. G. Vogt, D. Iuga, A. J. Edwards and S. P. Brown, Mol. Pharmaceutics, 2013, 10, 999–1007 CrossRef CAS PubMed.
- M. Grüne, R. Luxenhofer, D. Iuga, S. P. Brown and A. C. Pöppler, J. Mater. Chem. B, 2020, 8, 6827–6836 RSC.
- X. Lu, D. Skomski, K. C. Thompson, M. J. McNevin, W. Xu and Y. Su, Anal. Chem., 2019, 91, 6217–6224 CrossRef CAS.
- Y. L. Hong, G. N. M. Reddy and Y. Nishiyama, Solid State Nucl. Magn. Reson., 2020, 106, 101651 CrossRef CAS PubMed.
- C. M. Quinn, R. Zadorozhnyi, J. Struppe, I. V. Sergeyev, A. M. Gronenborn and T. Polenova, Anal. Chem., 2021, 93, 13029–13037 CrossRef CAS PubMed.
- T. Mizuno, T. Nemoto, M. Tansho, T. Shimizu, H. Ishii and K. Takegoshi, J. Am. Chem. Soc., 2006, 128, 9683–9686 CrossRef CAS PubMed.
- A. E. Aliev, S. E. Mann, D. Iuga, C. E. Hughes and K. D. M. Harris, J. Phys. Chem. A, 2011, 115, 5568–5578 CrossRef CAS PubMed.
- M. Schulz-Dobrick and I. Schnell, Cent. Eur. J. Chem., 2005, 3, 245–251 CAS.
- A. J. Rossini, J. Schlagnitweit, A. Lesage and L. Emsley, J. Magn. Reson., 2015, 259, 192–198 CrossRef CAS PubMed.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, P. Péchy, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2017, 139, 10055–10061 CrossRef CAS PubMed.
- P. Raval, R. M. Kennard, E. S. Vasileiadou, C. J. Dahlman, I. Spanopoulos, M. L. Chabinyc, M. Kanatzidis and G. N. M. Reddy, ACS Energy Lett., 2022, 7, 1534–1543 CrossRef CAS.
- S. Cavadini, S. Antonijevic, A. Lupulescu and G. Bodenhausen, J. Magn. Reson., 2006, 182, 168–172 CrossRef CAS PubMed.
- Z. Gan, J. P. Amoureux and J. Trébosc, Chem. Phys. Lett., 2007, 435, 163–169 CrossRef CAS.
- P. Raval, J. Trébosc, T. Pawlak, Y. Nishiyama, S. P. Brown and G. N. M. Reddy, Solid State Nucl. Magn. Reson., 2022, 120, 101808 CrossRef CAS PubMed.
- F. Schneider, L. Erisson, H. Beygi, M. Bradbury, O. Cohen-Barak, I. D. Grachev, S. Guzy, P. S. Loupe, M. Levi, M. McDonald, J. M. Savola, S. Papapetropoulos, W. G. Tracewell, M. Velinova and O. Spiegelstein, Br. J. Clin. Pharmacol., 2018, 84, 2422–2432 CrossRef CAS PubMed.
- S. K. Jain, A. B. Nielsen, M. Hiller, L. Handel, M. Ernst, H. Oschkinat, Ü. Akbey and N. C. Nielsen, Phys. Chem. Chem. Phys., 2014, 16, 2827–2830 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and NMR spectral analysis. See DOI: https://doi.org/10.1039/d2cc04585k |
| ‡ Equally contributed to this work. |
| § Interatomic distances are measured from periodic DFT geometry optimized crystal structure (ESI,† Experimental section). |
| ¶ C–D bond is shorter by 0.5 picometers compared to the C–H bond, and a shortening is expected for the N–D bond distances. |
| This journal is © The Royal Society of Chemistry 2022 |