
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
7-Azaindole breaks carboxylic acid dimers and simplifies VCD spectra analyses of natural products†
Corentin
Grassin
 ,
Ernesto
Santoro
and
Christian
Merten
*
,
Ernesto
Santoro
and
Christian
Merten
*
Ruhr-Universität Bochum, Organische Chemie 2, Stereochemistry and Chiroptical Spectroscopy, Universitätsstraße 150, 44801, Bochum, Germany. E-mail: christian.merten@ruhr-uni-bochum.de; Web: https://www.mertenlab.de
First published on 23rd September 2022
Abstract
The determination of absolute configurations of carboxylic acids by vibrational circular dichroism (VCD) spectroscopy is often complicated by self-aggregation and the subsequent need to compute the spectra of the aggregates. We show that 7-azaindole effectively breaks up these aggregates by stronger complemental hydrogen bonding to the COOH moiety, enabling drastic simplification and acceleration of VCD spectra calculations.
Vibrational circular dichroism (VCD) spectroscopy has grown into an established method for the determination of absolute configurations (ACs) of synthetic compounds1,2 as well as natural products.3,4 It measures the differential absorbance of left- and right-circular polarized infrared light by vibrational transitions and thus, unlike its UV/vis counterpart electronic circular dichroism (ECD), does not require dedicated chromophores as all chiral molecules are IR-active. The actual determination of the AC is achieved by comparison of the experimental VCD signatures with the computed VCD spectrum of the proposed enantiomer: if the experimental and computed spectra match, the AC of the sample is that of the computed structure; if experimental and computed VCD spectra are mirror images, the sample has the opposite AC of the computed one.
The computation of VCD spectra requires a comprehensive conformation analysis as missing important conformation may lead to significant mismatches with the experiments and thus a potential problem in AC assignments. Furthermore, as VCD spectroscopy is a vibrational spectroscopic technique, strong hydrogen bonding interactions with the solvent or other solutes can have an additional influence on the VCD spectral pattern. Solute-solvent interactions with H-bonding solvents and self-aggregation, such as dimerization of carboxylic acids, must explicitly be considered in the computations.5–7 This can increase the computational efforts enormously, as the conformational space increases drastically, and may make the analysis of the VCD spectrum of a complex natural product potentially not feasible anymore. In case of carboxylic acids, for instance, the theoretical number of structures to compute increases from n monomer conformers to (n2 + n)/2 dimeric structures!
In order to develop general rules or guidelines helping to determine whether solvation needs to be considered, we have recently begun to systematically investigate solvent effects on a variety of functional groups8–12 relevant for natural products research and beyond. For the analysis of the VCD spectra of carboxylic acids,8,9 we found solvation to be important but also rather simple to consider: hydrogen bond acceptor solvents interact with the O–H group of the carboxylic acid and placing one solvent molecule of ACN or DMSO near the O–H group typically suffices to model the solvent effect on the spectra. Due to their high boiling points, these strongly polar solvents are often not the ideal solvents for VCD studies on natural products, as their removal may require some heating which can subsequently cause undesired rearrangement reactions of the often quite complex structures. When turning to weakly polar solvents, such as CDCl3, dimerization can be expected. Experimentally, aggregation can be prevented to a certain degree by choosing low enough concentrations for the measurements. Nonetheless, only after the experiment or after the computational analysis of the monomeric carboxylic acids are already done, it becomes clear whether dimerization has to be considered. A simplification of dimer computations can be achieved by using formic or acetic acid as truncated second acid models in the structure, but this approach may lead to artificial signals in the computed IR and potentially also in the VCD spectrum.5,8
Avoiding aggregation from the beginning usually requires chemical modification of the carboxylic acid, for instance by transforming it into a carboxylate salt or a methyl ester.13 While certainly being very efficient from the computational perspective, this chemical modification method has the same drawback as the Mosher ester approach in NMR spectroscopy: the sample cannot be used for any further experiments without notable loss of material. Considering that the use of VCD spectroscopy in the field of natural products is often already complicated due to insufficient amounts of sample material available for the measurement, such approaches are not generally applicable.
In light of the above-mentioned issues, we were seeking for an additive, that can be added in equimolar ratios directly to a carboxylic acid solution, that preferentially breaks carboxylic acid dimers instead of forming homodimers on its own, and that is subsequently easy to remove to avoid loss of precious sample material. One may immediately think of simply deprotonating the carboxylic acid with an amine. However, besides the fact that deprotonation might not be complete, simply selecting an arbitrary amine for this purpose comes with another major drawback for the analysis: the resulting ammonium and carboxylate ions could remain as contact ion pair, they could become separated and solvated, or there could be a mixture of these states present in solution. Hence, the structural modelling required to simulate the VCD spectrum of the sample would actually become even more complicated that for a homodimer. In this study, we introduce the structurally complemental base 7-azaindole (7AI, Scheme 1) as the ideal breaker of carboxylic acid homodimers. It provides a perfectly complemental structure that can interact through its basic site with the O–H and through the N–H with the carbonyl functionality of the carboxylic acid. Thereby it binds to the COOH moiety in a double hydrogen bonded manner just like another carboxylic acid would, but it binds stronger due to its basic character.
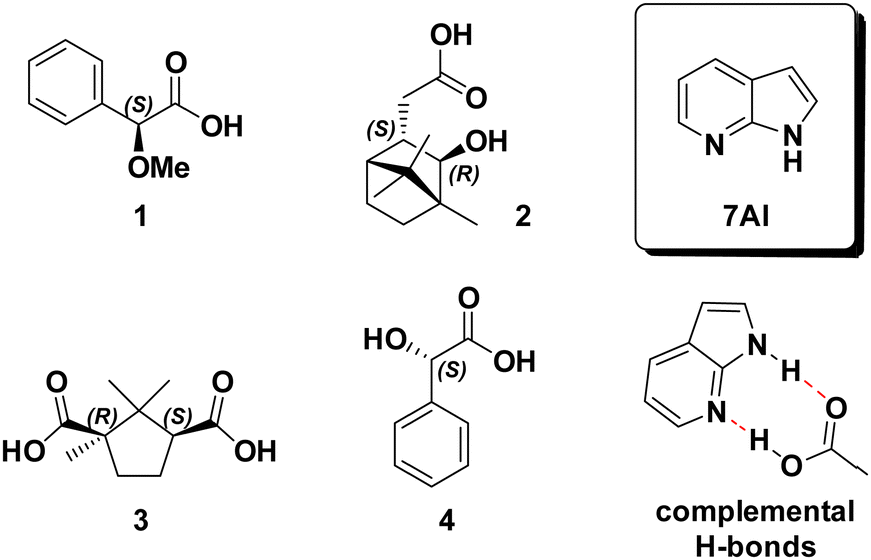 | ||
| Scheme 1 Structure of 7-azaindole (7AI) and of the four investigated carboxylic acids 1–4. The hydrogen bonding sides of 7AI perfectly match those of the COOH group. | ||
In a previous study, we investigated α-methoxyphenyl propionic acid 1 as model substance for solvation studies on carboxylic acids.9 In this context, we also recorded and analysed 1 in chloroform-d1 solution. In the concentration range regularly employed for VCD spectroscopic determinations of ACs (∼50–100 mg mL−1), 1 was shown to predominantly exist as dimeric species. This was unambiguously confirmed by comparing the experimental spectra with computed spectra of (1)2 (cf.Fig. 1). Anticipating that 7AI would effectively break dimers of a carboxylic acid, we prepared an equimolar mixture of 7AI and 1 at the same concentration which previously led to strong dimer formation (0.3 M). The direct comparison with the IR spectrum of pure 1 reveals some clear changes: the band of the carbonyl stretching mode at ∼1730 cm−1 decreases strongly in intensity and broadens notably. In particular in the range below 1300 cm−1, new bands arise that can be assigned to vibrational modes of 7AI (cf. ESI† file, Fig. S1). The most pronounced change in the VCD spectrum of the mixture is the disappearance of the strong (+/−/−)-couplet in the carbonyl region, while changes in the range below 1500 cm−1 are rather small and subtle.
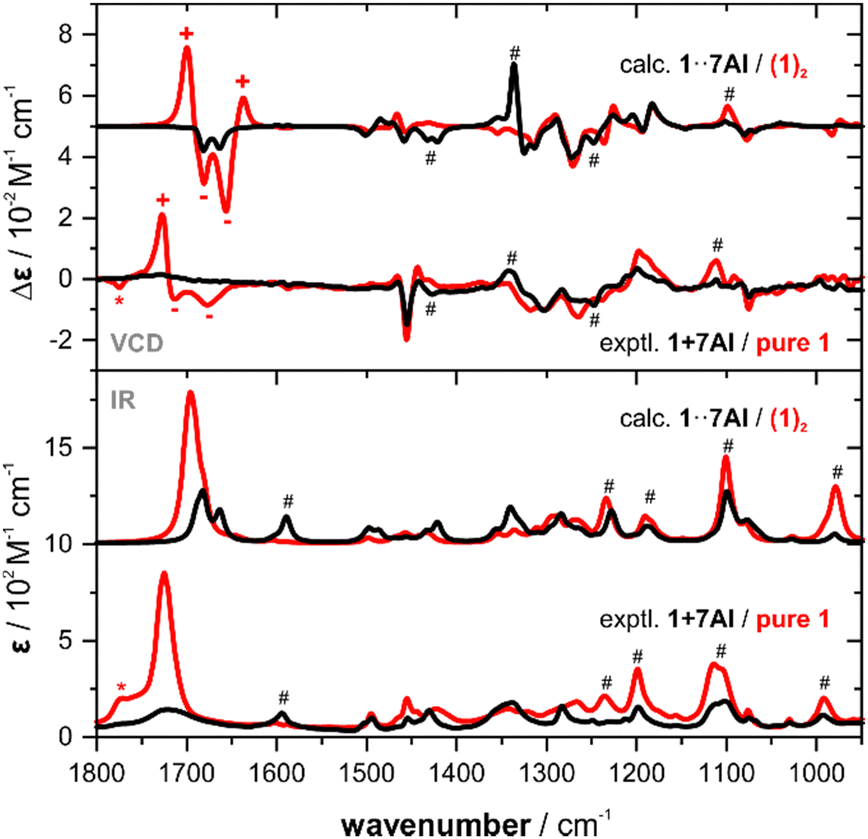 | ||
| Fig. 1 Comparison of the experimental IR and VCD spectra of 1 and an equimolar mixture of 1 with 7AI (0.3 M, 100 μm optical pathlength, CDCl3) with the corresponding computed spectra. The asterisk marks a residual band of monomeric 1,9 hashtags mark characteristic changes in the experimental spectra that are reproduced in the simulated spectra. A direct overlap of the experimental and computed spectra of 1 + 7AI is provided in the ESI† Fig. S2. | ||
In order to further analyse the observed spectral changes and to correlate them to a solution phase structure, we carried out spectra calculations. To this end, we first retrieved the six monomer conformations of 1, their relative energies and Boltzmann weights from our previous study. Based on these structures, hydrogen bonded clusters 1·7AI (cf.Scheme 1) were built by placing 7AI near the carboxylic acid moiety. After geometry optimization and vibrational spectra calculations, we thus obtained six cluster structures denoted 1·7AI-c1 to -c6. The conformer numbering c1 to c6 refers to the original conformer order obtained for 1, as the conformation of the carboxylic acid was indeed almost unaffected by the coordination to 7AI. Likewise, the conformer energies of 1 and 1·7AI (Table 1, Table S1 and S2, ESI†) revealed that the conformational distribution was also not affected by binding to 7AI.
| Monomeric 1 | Hydrogen bonded cluster 1·7AI | ||||
|---|---|---|---|---|---|
| ΔEZPC | χ(ΔE) | ΔEZPC | χ(ΔE) | ||
| 1-c1 | 0.00 | 44.0 | 1·7AI-c1 | 0.00 | 44.9 |
| 1-c2 | 0.39 | 22.9 | 1·7AI-c2 | 0.40 | 22.7 |
| 1-c3 | 0.44 | 20.8 | 1·7AI-c3 | 0.43 | 21.5 |
| 1-c4 | 0.79 | 11.6 | 1·7AI-c4 | 0.87 | 10.3 |
| 1-c5 | 2.59 | 0.6 | 1·7AI-c5 | 2.77 | 0.4 |
| 1-c6 | 3.58 | 0.1 | 1·7AI-c6 | 3.47 | 0.1 |
The IR and VCD spectra of the six conformers of 1·7AI were subsequently used to compute the Boltzmann-averaged spectra of the hydrogen bonded complex. The direct overlap with the computed spectra of the dimer presented in Fig. 1 allows for a straightforward confirmation that the experimentally observed spectral changes are overall very nicely captured by the simulation. The disappearance of the strong carbonyl feature is also well reproduced; note that the experimental IR and VCD bands in the carbonyl region are much broader and thus appear weaker than those in the simulated spectra. Likewise, changes in the experimental VCD signatures in the range below 1500 cm−1 can also be found in the computed spectra. For instance, the shift and slight intensity gain for the (+/−) signature at 1340 cm−1 is predicted by the calculations, although again sharper and thus more intense due to the uniform line broadening.
The addition of one equivalent of 7AI to 1 lead to the formation of a one-to-one hydrogen bonded cluster (cf.Fig. 2) and 7AI could easily be removed by column chromatography or separation (cf. ESI† for details). Experiments with excess amounts of 7AI gave essentially the same VCD response. Similar results were also obtained for the addition of 7AI to 2-phenylpropionic acid8 (cf. ESI,† Fig. S3 and Tables S6 and S7). Hence, as excess of 7AI did not lead to deprotonation of carboxylic acids, we hypothesized that intramolecular hydrogen bonds, such as the interaction between the COOH and OH group in isoborneolacetic acid 2, may also remain intact. Therefore, analogously to 1, we recorded the IR and VCD spectra of 2 and an equimolar mixture of 2 and 7AI to evaluate this hypothesis. The two sets of experiments revealed mostly differences in the IR spectra, which are almost entirely associated with vibrational modes of 7AI. Again, the decrease and broadening of the carbonyl stretching region is the most indicative sign for a change in the solution phase structure of 2. Differences between the VCD spectra are even more difficult to identify. Addition of 1.5 or 2 eq. of 7AI did not lead to any further changes of the VCD signatures (cf. ESI,† Fig. S4).
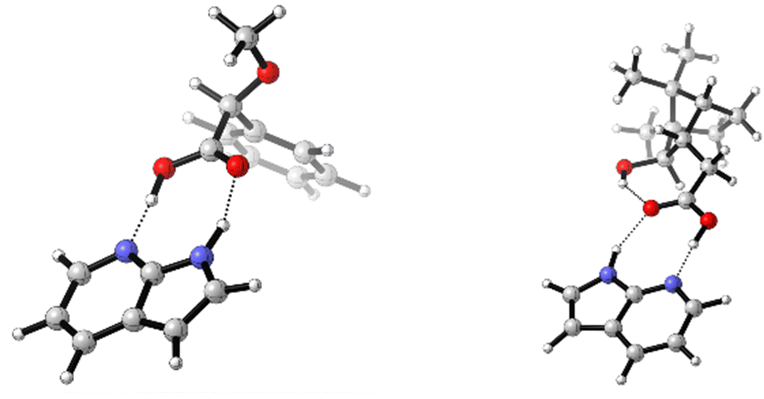 | ||
| Fig. 2 Structure of the lowest energy conformers 1·7AI-c1 and 2·7AI-c1. | ||
A systematic conformational analysis of 2, which evaluated three rotable bonds, gave 15 monomer conformations (cf. Table S3, ESI† for geometries). Interestingly, four of these conformers had a cis-conformation of the COOH moiety with 2-c2 even showing an intramolecular hydrogen bond pointing from COOH to the hydroxy group. The first five lowest energy conformers with trans-COOH conformation (i.e. leaving out 2-c2) were systematically paired giving 15 dimer structures (Table S4, ESI†). Likewise, 7AI-complexes with all trans-COOH conformers were optimized (Table S5, ESI†). Comparing the resulting sets of structures, it is noted that the conformational distribution of 2 is again not particularly affected by hydrogen bond formation with either another molecule of 2 or with 7AI: the main conformer 2-c1 (76% population) remains the most stable in the dimer (dimer 2-c1–c1 has 65% population) and in the heterodimer 2·7AI (2·7AI-c1 with 83% population; Fig. 2).
As observed for the experimental spectra, the computed VCD spectra of (2)2 and 2·7AI are also quite similar (Fig. 3). Nonetheless, most experimentally observed differences are found in the predicted VCD spectra. Likewise, the computed IR spectra resemble most of the differences seen in the experiment, although some bands appear to be sharper due to the uniform line broadening. Finally, it should be noted that the computed spectra of monomeric 2 do not match as nicely with the experiment as those of (2)2, thus confirming that dimerization takes place in CDCl3 solution (Fig. S5, ESI†). From the VCD spectra analysis of pure 2 and the mixtures of 2 with 7AI it can be concluded that 7AI indeed does not break the intramolecular hydrogen bond even when added in excess. The complemental hydrogen bonding pattern leads to a favourable formation of either heterodimers 2·7AI or, in case of excess of 7AI, to the formation of VCD-inactive homodimers (7AI)2.
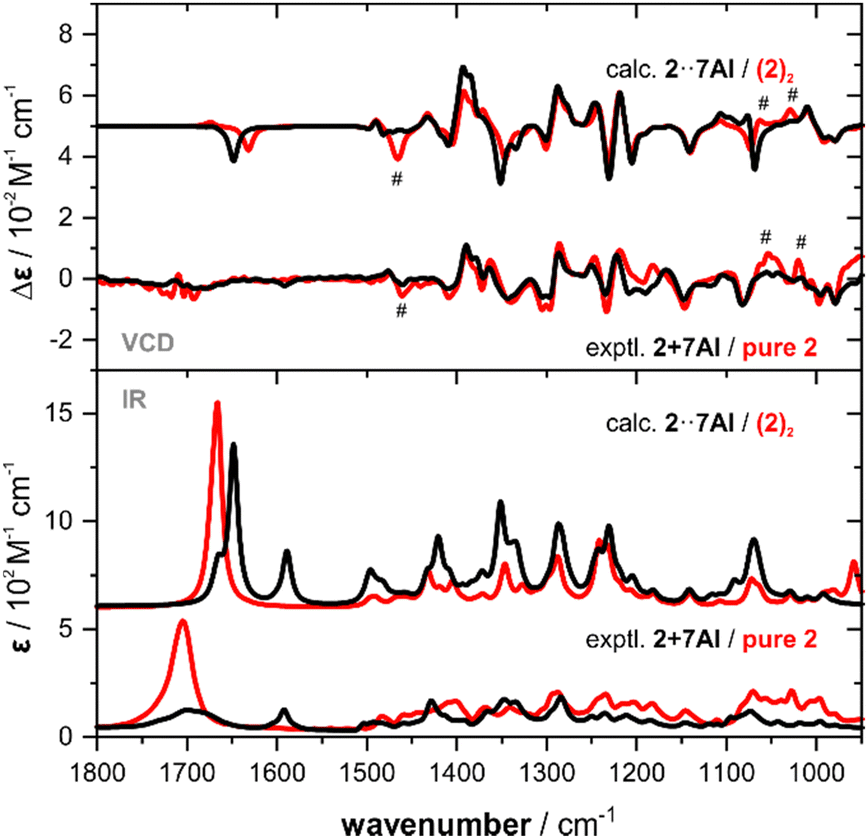 | ||
| Fig. 3 Comparison of the experimental IR and VCD spectra of 2 and an equimolar mixture of 2 with 7AI (0.19 M, 100 μm optical pathlength, CDCl3) with the corresponding computed spectra. Hashtags mark characteristic changes in the experimental spectra that are reproduced in the simulated spectra. | ||
Occasionally, carboxylic acids are not soluble in weakly polar media such as chloroform(-d1) or dichloromethane(-d2), which are preferred solvents for VCD spectroscopic analysis. One such example is camphoric acid 3, which only dissolves in hydrogen bonding solvents. However, as mentioned before, this may complicate the analysis when solvation needs to be considered explicitly in the spectra predictions.8,9 In a mixture with two equivalents of 7AI, the dicarboxylic acid 3 dissolved in CDCl3 solution and we successfully recorded its IR and VCD spectra (cf.Fig. 4).
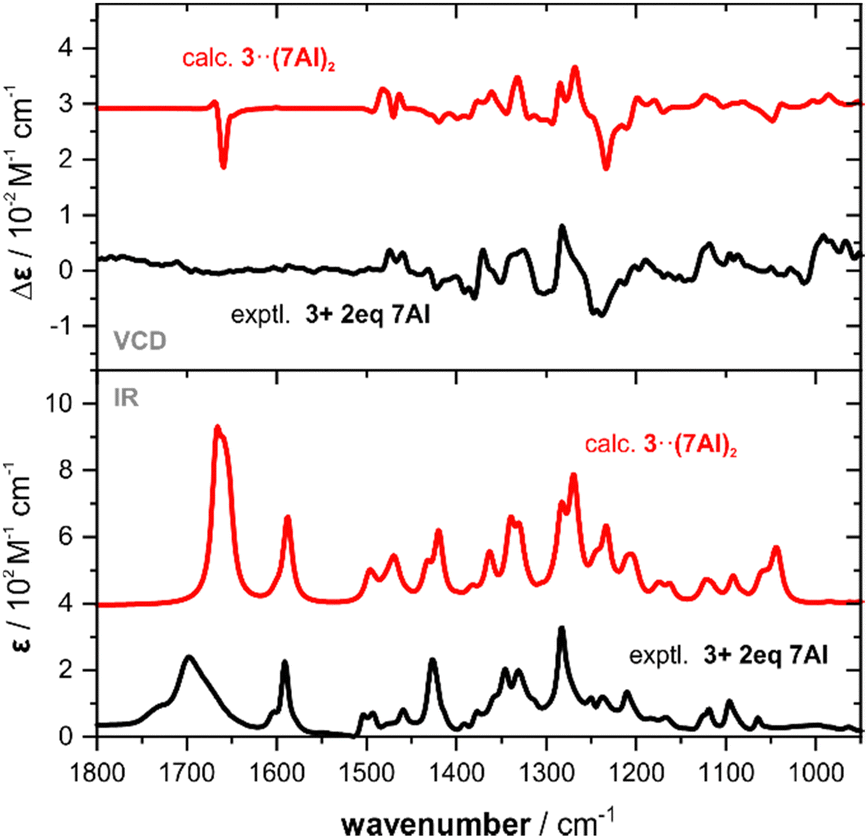 | ||
| Fig. 4 Comparison of the experimental IR and VCD spectra of 3 and an equimolar mixture of 3 with 7AI (0.25 M, 100 μm optical pathlength, CDCl3) with the corresponding computed spectra. Intensities of the heterotrimer spectra are scaled by a factor of 0.5 for better comparison. | ||
For the spectra simulations, we considered 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 heterotrimeric structures 7AI·3·7AI (cf. Fig. S7, ESI†). The six conformers of 3, that one can find when considering the rotation of the trans-COOH moieties and ring puckering, again do not change much in their energetic order when forming the trimeric cluster (c4 and c5 change the order) and about 90% of the Boltzmann weight remains on the first three conformers (cf. Tables S8 and S9, ESI†). The simulated spectra of 3·(7AI)2 are found to match nicely with the experimental spectra (cf.Fig. 4) and all VCD spectral features are captured. Solely, as already observed for the other examples, the carbonyl band appear stronger due to the narrow simulated line broadening.
:2 heterotrimeric structures 7AI·3·7AI (cf. Fig. S7, ESI†). The six conformers of 3, that one can find when considering the rotation of the trans-COOH moieties and ring puckering, again do not change much in their energetic order when forming the trimeric cluster (c4 and c5 change the order) and about 90% of the Boltzmann weight remains on the first three conformers (cf. Tables S8 and S9, ESI†). The simulated spectra of 3·(7AI)2 are found to match nicely with the experimental spectra (cf.Fig. 4) and all VCD spectral features are captured. Solely, as already observed for the other examples, the carbonyl band appear stronger due to the narrow simulated line broadening.
Finally, we note that the solubilizer properties of 7AI also do not contradict the findings on the stability of intramolecular hydrogen bonds. In fact, mandelic acid 4, which is insoluble in chloroform(-d1), readily dissolves upon addition of one equivalent of 7AI and the corresponding computed spectra of 4·7AI would again allow a straightforward determination of the AC (cf. Fig. S6 and Tables S10 and S11, ESI†).
In summary of this study, we introduced a very simple concept to avoid computing dimeric structures of carboxylic acids for VCD spectra analysis. The procedure is experimentally simple, as it just requires the addition of one equivalent of 7AI per carboxylic acid moiety. The 7AI breaks homodimers of the COOH moieties and forms heterodimeric and heterotrimeric structures instead. Furthermore, the procedure is experimentally safe, as 7AI preferentially binds to the COOH moiety and does not lead to the deprotonation of OH groups. Hence, 7AI can safely be added to even more complex carboxylic acid compounds prior to the measurement without needing to worry about an initiation of rearrangement reactions or alike. The main simplification of the analysis introduced by this auxiliary becomes obvious when comparing the number of computed structures: the explicit consideration of 7AI in the calculations does not increase the number of conformers that have to be considered, while homodimerization of carboxylic acids results in (n2 + n)/2 dimeric structures. Consequently, 7AI can be considered in the conformational search from the beginning (or easily incorporated into the structures afterwards). We thus expect 7AI to largely simplify future VCD spectroscopic characterizations of natural products with COOH moieties or even to enable them at all.
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy (EXC-2033, project number 390677874) and by the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 801459. C. M. acknowledges support through the DFG's Heisenberg program (ME 4267/5-1, project no. 418661145).
Conflicts of interest
There are no conflicts to declare.References
- C. Merten, T. P. Golub and N. M. Kreienborg, J. Org. Chem., 2019, 84, 8797–8814 CrossRef CAS.
- P. J. Stephens, F. J. Devlin and J. R. Cheeseman, VCD Spectroscopy for Organic Chemists, CRC Press, 2012 Search PubMed.
- J. M. Batista Jr., E. W. Blanch and V. da Silva Bolzani, Nat. Prod. Rep., 2015, 32, 1280–1302 RSC.
- P. L. Polavarapu and E. Santoro, Nat. Prod. Rep., 2020, 37, 1661–1699 RSC.
- J. M. Batista, A. N. L. Batista, D. Rinaldo, W. Vilegas, D. L. Ambrósio, R. M. B. Cicarelli, V. S. Bolzani, M. J. Kato, L. A. Nafie, S. N. López and M. Furlan, J. Nat. Prod., 2011, 74, 1154–1160 CrossRef CAS PubMed.
- R. O. P. Gema, M. G. Manuel, M. L. Fernando and L. G. J. Jesús, Chem. – Asian J., 2016, 11, 1798–1803 CrossRef.
- S. Gobi, E. Vass, G. Magyarfalvi and G. Tarczay, Phys. Chem. Chem. Phys., 2011, 13, 13972–13984 RSC.
- K. Bünnemann, C. H. Pollok and C. Merten, J. Phys. Chem. B, 2018, 122, 8056–8064 CrossRef PubMed.
- K. Bünnemann and C. Merten, Phys. Chem. Chem. Phys., 2017, 19, 18948–18956 RSC.
- L. Weirich, J. Magalhaes de Oliveira and C. Merten, Phys. Chem. Chem. Phys., 2020, 22, 1525–1533 RSC.
- L. Weirich, K. Blanke and C. Merten, Phys. Chem. Chem. Phys., 2020, 22, 12515–12523 RSC.
- L. Weirich and C. Merten, Phys. Chem. Chem. Phys., 2019, 21, 13494–13503 RSC.
- P. L. Polavarapu, E. A. Donahue, K. C. Hammer, V. Raghavan, G. Shanmugam, I. Ibnusaud, D. S. Nair, C. Gopinath and D. Habel, J. Nat. Prod., 2012, 75, 1441–1450 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc04433a |
| This journal is © The Royal Society of Chemistry 2022 |