
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilicon–nitrogen bond formation via dealkynative coupling of amines with bis(trimethylsilyl)acetylene mediated by KHMDS†
Krzysztof
Kuciński
 * and
Grzegorz
Hreczycho
* and
Grzegorz
Hreczycho
Faculty of Chemistry, Adam Mickiewicz University in Poznań, Ul. Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: kucinski.k@amu.edu.pl
First published on 13th September 2022
Abstract
The catalytic synthesis of silylamines mediated by s- and p-block catalysts is largely underdeveloped. Herein, commercially available potassium bis(trimethylsilyl)amide serves as an efficient alternative to transition metal complexes. N–H/Si–C dealkynative coupling was achieved by means of user-friendly main-group catalysis with ample substrate scope and high chemoselectivity.
Silylamines are prevalent in organic chemistry, so new methods are needed for their synthesis.1–4 Indeed, these compounds wherein the silyl moiety is directly bonded to nitrogen have found broad applications, including their utilization as bases,5 silylating agents (e.g., needed for the protection of reactive groups such as OH),6–8 and ligands for several complexes (Fig. 1a).9–15 Aminosilanes can be readily accessed via well-developed stoichiometric methods. Here, the Si–N bond is formed by aminolysis of chlorosilanes with amines (Fig. 1b).16 Because of the inconvenient nature of these processes (e.g., high moisture sensitivity of substrates, generation of salts and other acidic wastes, etc.), researchers have tried to develop catalytic alternatives. These methods can be generally divided into hydrosilylation of imines17 and coupling between amines with different silylating agents (hydrosilanes, vinylsilanes, and ethynylsilanes). All reactions have good or high atom efficiency, producing no waste or easy-to-separate byproducts such as dihydrogen, ethylene, or acetylene. The dehydrocoupling between amines and hydrosilanes seems to be the most attractive route, due to the formation of H2 as the by-product (Fig. 1c).18–36 Unfortunately, this approach is completely impractical for the preparation of a highly important trimethylsilyl-protecting group, due to the pyrophoricity of gaseous Me3SiH. Moreover, the control of the chemoselectivity for Si–H/N–H dehydrocoupling has proved to be challenging due to the possible formation of several products including polymeric silazanes and a mixture of mono- and disilylated amines. On the other hand, the use of vinylsilanes within the dealkenative strategy gives high chemoselectivity for the formation of the Si–N monocoupling product (Fig. 1d).37
 | ||
| Fig. 1 Context of the investigation. | ||
However, other features of this process (e.g., expensive Ru catalyst, long reaction time, possible homocoupling of vinylsilane, etc.) dramatically reduce its potential. Finally, amines can be N–silylated by using silylacetylenes under basic conditions. To the best of our knowledge, there is only one example of such an atypical synthetic strategy.38 Baba et al. showed that MgO or KNH2 loaded on alumina, can be used as catalysts in dealkynative coupling between amines and silylacetylenes (Fig. 1e). Notably, this strategy is not without its own disadvantages, including the use of dimethylformamide as the solvent and a long reaction time (20 h). Moreover, the authors have reported only one product.
Sustainable and eco-friendly synthetic approaches proceeded by main-group catalysis have gained recent significant attention.39–44 On the basis of our recent success in activating silylacetylenes under sustainable catalysis,45–48 we reasoned that an appropriate catalytic manifold could provide an efficient platform to generate diversified libraries of trimethylsilylated amines. In this communication, we report on the catalytic silylation of primary amines with bis(trimethylsilyl)acetylene (BTMSA) for the construction of N–Si bonds via dealkynative coupling, by using potassium bis(trimethylsilylamide) as the catalyst (Fig. 1f).
In optimization studies, summarized in Table 1 (Table S1 in ESI†), we investigated catalytic N–H trimethylsilylation of 3-methylaniline (1a). Initial success was achieved using bis(trimethylsilyl)acetylene (2a) as the silylating agent, and KHMDS as the catalyst. Using MeCN as a solvent, this main-group catalytic combination afforded the desired product 3a in 97% yield (entry 1). Control experiments showed that other main-group mediators were also active in this transformation (entries 7–9), but gave inferior results. Particularly noteworthy is a very good conversion of 1a in the presence of 5 mol% KOH (90% at r.t., and 95% at 50 °C). However, we decided to continue our work with KHMDS, due to less problematic isolation and better yields of the final products. A catalyst-free attempt was also carried out and proved the essential role of the main-group catalysis and confirmed no leaching of the alkali species from the glassware, which could act as potential co-catalysts (entry 2, the results verified after 24 h).50 The reaction can also be performed under an air atmosphere, although the conversion was slightly lower (entry 3). The use of MeCN as a solvent was critical, as demonstrated by the lack of conversion observed in toluene and dioxane (entries 12 and 13) and the lower yield (75%) obtained in THF (entry 11). These results suggest that acetonitrile might play a more important role in the reaction process, possibly acting as the stabilizer of Si-containing intermediates. Next, an attempt to replace the silylating source with trimethylsilyl-acetylene gave an inferior result (entry 4).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Conversion of 1ab [%] |
| a Reaction conditions: 1a (1 mmol), 2a (1 mmol), under argon atmosphere. b Conversion determined via GC, with n-dodecane as the internal standard. c Isolated yield in parentheses. d In a brand-new set of equipment, to exclude the influence of any transition metal impurities.49 e After 2 h and 10 h. f 2 eq. of trimethylsilylacetylene. | ||
| 1 | No change | 99 (97)c |
| 2 | No catalyst | 0d |
| 3 | Under air atmosphere | 97 |
| 4 | Trimethylsilylacetylene instead of 2a | 95ef |
| 5 | Trimethyl(phenylethynyl)silane instead of 2a | 16e |
| 6 | 0.5 eq. of BTMSA | 85 |
| 8 | 1.5 mol% of KHMDS | 95 |
| 8 | 3 mol% of KOH | 85 |
| 9 | 5 mol% of KOH (r.t./50 °C) | 90/95 (92)c |
| 10 | 3 mol% of t-BuOK | 94 |
| 11 | 3 mol% of KF | 0 |
| 12 | In tetrahydrofuran | 75 |
| 13 | In toluene | 0 |

We next studied the scope with respect to primary aromatic amines (Scheme 1). A wide range of N-silylated anilines were accessed in high yields and with perfect chemoselectivity, leading to monosilylated derivatives exclusively (3a–3n). As an initial example 3a, anilines bearing electron-donating alkyl groups were readily silylated (3c–3f).
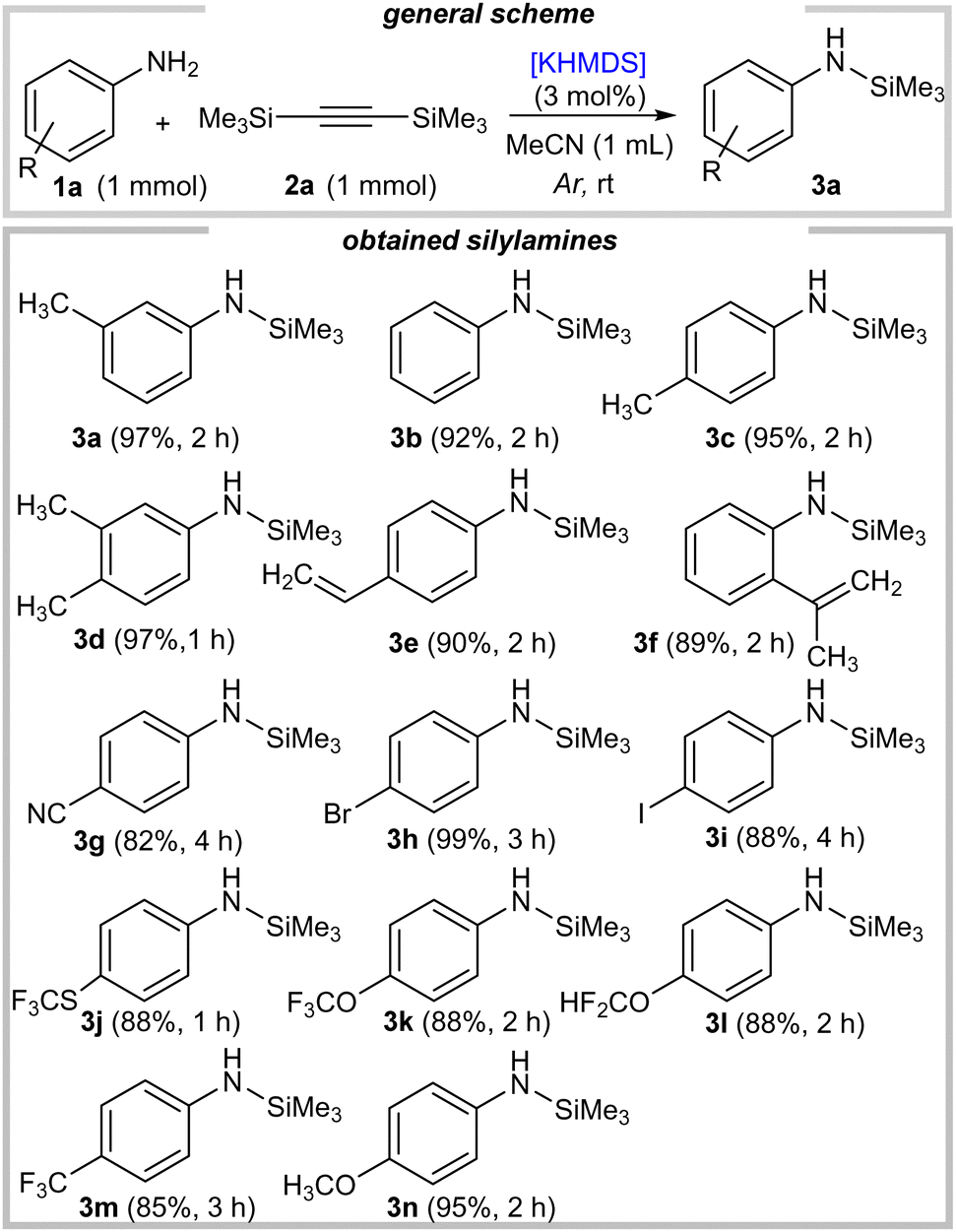 | ||
| Scheme 1 Substrate scope for silylation of variously substituted anilines. | ||
Additionally, electron-rich methoxy substituted reagent 1n also gave the corresponding product 3n (95% yield). Gratifyingly, halo-substituted anilines were readily adopted in this protocol (3h–3i, 88–99% yield; in the case of 3i, there was incomplete conversion ∼ 93%), as were electron-deficient ones bearing fluorinated functionalities (3j–3m, 85–88% yield). Moreover, the synthetically useful cyano derivative 1g was also well tolerated, and led to silylated product 3g in very good yield (82%). Encouraged by these results, we then investigated the use of heterocyclic primary amines, which are synthetically useful and biorelevant scaffolds (Scheme 2).
 | ||
| Scheme 2 Substrate scope for silylation of amine-substituted heterocycles. | ||
All of them afforded the expected products in excellent yields (5a–5f, 91–99%), including amine-substituted pyridines, pyrimidine, pyrazine, quinoline, and benzothiazole. All these examples highlight both the electronic generality of this method and its tolerance for typically existing organic motifs, showcasing the unique robustness and versatility of our strategy. Notably, our initial studies ruled out the efficient silylation via dealkynative coupling (please see ref. 47 and 48). The main problem was related to the acidity of the amine. When we used more basic, aliphatic amines we did not observe any product, whilst the use of N-methylaniline gave only traces of the product. Finally, the use of primary amines with more acidic -NH2 protons showed an excellent conversion to their silylated forms.
Finally, our protocol was scaled up to a 10 mmol scale yielding 92% (2.16 g) of the silylated product 3m (Scheme 3). This once again makes it clear that the proposed methodology has a significant application potential.
 | ||
| Scheme 3 Scaled-up synthesis of 3m. | ||
Given the fact of our previous studies concerning O–H and sp C–H silylation in the presence of KHMDS,47,48 anilines with hydroxyl and ethynyl moieties were also examined (Scheme 4). For this purpose, an equimolar combination of BTMSA and bifunctional amines was subjected under our reaction conditions. In the case of 4-aminophenol (1o), O-silylated derivative 3o was obtained exclusively (99%). Further addition of BTMSA gave a hard-to-separate mixture of O- and N-silylated products. On the other hand, 3-ethynylaniline (1p) led efficiently to C,N-silylated product 3p (88%), in the presence of 2.0 eq. of BTMSA (compared to an observed mixture of silylation products when 1.0 eq. of 2a was applied).
 | ||
| Scheme 4 Competition experiments. | ||
To gain some mechanistic insights into this main-group catalysis, we conducted preliminary experiments. The trimethylsilylation was performed in the presence of a typical radical scavenger such as TEMPO (100 mol%), giving the desired product (with almost the same efficiency), thereby implying that radical pathways were likely not operative (see ESI†).51 We next evaluated the role of the potassium cation. Therefore, the reaction of 1m with 2a was performed in the presence of chelating agent 18-crown-6, as well as metal scavenger Quadra-Pure®TU (for both experiments see ESI†). As a result, again, the desired product was obtained with almost the same efficiency, thereby suggesting that the metal ion does not play any decisive role in our process. In general, as already mentioned, the acidity of the amine is a very important factor. It somehow confirms the importance of a deprotonation step. A stoichiometric reaction between 1m and KHMDS confirmed (by 1H NMR) the disappearance of protons from the NH2 group (for details see ESI†).
On the basis of our experimental results and previous literature, a plausible catalytic cycle is presented for the N–H trimethylsilylation (Fig. 2).
 | ||
| Fig. 2 The plausible mechanism. | ||
In summary, we have reported on a very efficient protocol for catalytic N–H silylation of aromatic primary amines under main-group catalysis. Here, a commercially available KHMDS enabled a dealkynative coupling with ample scope. Considering the combination of desirable features, such as operational simplicity, high chemoselectivity, good atom economy, benign reaction conditions, low cost of the reagents, and their commercial availability, this reaction system is expected to provide a promising alternative to existing methodologies. Moreover, the mechanistic studies provided strong support for the deprotonation step. In a broader context, this interesting example of dealkynative coupling might inspire the design of novel atom-economical reactions.
This work was supported by a National Science Centre Grant UMO-2018/30/E/ST5/00045 (GH).
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. B. Reuter, K. Hageman and R. Waterman, Chem. – Eur. J., 2021, 27, 3251–3261 CrossRef CAS PubMed
.
- V. Verma, A. Koperniku, P. M. Edwards and L. L. Schafer, Chem. Commun., 2022, 58, 9174–9189 RSC
- K. Kuciński, H. Stachowiak, D. Lewandowski, M. Gruszczyński, P. Lampasiak and G. Hreczycho, J. Organomet. Chem., 2022, 961, 122127 CrossRef
- K. Kuciński and G. Hreczycho, ChemCatChem, 2017, 9, 1868–1885 CrossRef
-
D. A. Armitage, The Silicon–Heteroatom Bond, John Wiley & Sons, Ltd, 1991, pp. 365–446 Search PubMed
- S. T. Kadam and S. S. Kim, Green Chem., 2010, 12, 94–98 RSC
- K. Kuciński and G. Hreczycho, Eur. J. Org. Chem., 2016, 4577–4585 CrossRef
- K. Kuciński and G. Hreczycho, ChemSusChem, 2019, 12, 1043–1048 CrossRef
- F. T. Edelmann, Coord. Chem. Rev., 1994, 137, 403–481 CrossRef
- M. Westerhausen, Coord. Chem. Rev., 1998, 176, 157–210 CrossRef CAS
- M. P. Coles, Coord. Chem. Rev., 2015, 297–298, 2–23 CrossRef CAS
- P. Schüler, H. Görls, M. Westerhausen and S. Krieck, Dalton Trans., 2019, 48, 8966–8975 RSC
- E. Le Coz, J. Hammoud, T. Roisnel, M. Cordier, V. Dorcet, S. Kahlal, J.-F. Carpentier, J.-Y. Saillard and Y. Sarazin, Chem. – Eur. J., 2021, 27, 11966–11982 CrossRef CAS
- A. Logallo and E. Hevia, Chimia, 2022, 76, 336 CrossRef CAS
- I. V. Lapshin, A. V. Cherkasov, K. A. Lyssenko, G. K. Fukin and A. A. Trifonov, Inorg. Chem., 2022, 61, 9147–9161 CrossRef CAS PubMed
- R. Fessenden and J. S. Fessenden, Chem. Rev., 1961, 61, 361–388 CrossRef CAS
- B. Li, J.-B. Sortais and C. Darcel, RSC Adv., 2016, 6, 57603–57625 RSC
- T. T. Nguyen, T. K. Mukhopadhyay, S. N. MacMillan, M. T. Janicke and R. J. Trovitch, ACS Sustainable Chem. Eng., 2022, 10, 4218–4226 CrossRef CAS
- L. Wirtz, J. Lambert, B. Morgenstern and A. Schäfer, Organometallics, 2021, 40, 2108–2117 CrossRef CAS
- Q. Bonnin, T. Edlová, E. D. Sosa Carrizo, P. Fleurat-Lessard, S. Brandès, H. Cattey, P. Richard, P. Le Gendre and A. T. Normand, Chem. – Eur. J., 2021, 27, 18175–18187 CrossRef CAS
- D. Gasperini, A. K. King, N. T. Coles, M. F. Mahon and R. L. Webster, ACS Catal., 2020, 10, 6102–6112 CrossRef CAS
- M. B. Reuter, M. P. Cibuzar, J. Hammerton and R. Waterman, Dalton Trans., 2020, 49, 2972–2978 RSC
- M. Rauch, R. C. Roberts and G. Parkin, Inorg. Chim. Acta, 2019, 494, 271–279 CrossRef CAS
- F. Palumbo, S. Rohrbach, T. Tuttle and J. A. Murphy, Helv. Chim. Acta, 2019, 102, e1900235 CrossRef CAS
- L. J. Morris, G. R. Whittell, J.-C. Eloi, M. F. Mahon, F. Marken, I. Manners and M. S. Hill, Organometallics, 2019, 38, 3629–3648 CrossRef CAS
- N. Li and B.-T. Guan, Eur. J. Inorg. Chem., 2019, 2231–2235 CrossRef CAS
- M. P. Cibuzar and R. Waterman, Organometallics, 2018, 37, 4395–4401 CrossRef CAS
- K. A. Erickson, M. P. Cibuzar, N. T. Mucha and R. Waterman, Dalton Trans., 2018, 47, 2138–2142 RSC
- P. Ríos, M. Roselló-Merino, O. Rivada-Wheelaghan, J. Borge, J. López-Serrano and S. Conejero, Chem. Commun., 2018, 54, 619–622 RSC
- N. V. Forosenko, I. V. Basalov, A. V. Cherkasov, G. K. Fukin, E. S. Shubina and A. A. Trifonov, Dalton Trans., 2018, 47, 12570–12581 RSC
- A. Baishya, T. Peddarao and S. Nembenna, Dalton Trans., 2017, 46, 5880–5887 RSC
- N. Li and B.-T. Guan, Adv. Synth. Catal., 2017, 359, 3526–3531 CrossRef CAS
- C. Bellini, T. Roisnel, J.-F. Carpentier, S. Tobisch and Y. Sarazin, Chem. – Eur. J., 2016, 22, 15733–15743 CrossRef CAS PubMed
- S. Anga, Y. Sarazin, J. F. Carpentier and T. K. Panda, ChemCatChem, 2016, 8, 1373–1378 CrossRef CAS
- J. F. Dunne, S. R. Neal, J. Engelkemier, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2011, 133, 16782–16785 CrossRef CAS PubMed
- H. Q. Liu and J. F. Harrod, Organometallics, 1992, 11, 822–827 CrossRef CAS
- B. Marciniec, S. Kostera, P. Pawluc and B. Wyrzykiewicz, Dalton Trans., 2015, 44, 782–786 RSC
- T. Baba, Y. Kawanami, H. Yuasa and S. Yoshida, Catal. Lett., 2003, 91, 31–34 CrossRef CAS
- H. Gao, A. Battley and E. M. Leitao, Chem. Commun., 2022, 58, 7451–7465 RSC
- K. Kuciński, H. Stachowiak-Dłużyńska and G. Hreczycho, Coord. Chem. Rev., 2022, 459, 214456 CrossRef
- M. M. D. Roy, A. A. Omaña, A. S. S. Wilson, M. S. Hill, S. Aldridge and E. Rivard, Chem. Rev., 2021, 121, 12784–12965 CrossRef
- M. Magre, M. Szewczyk and M. Rueping, Chem. Rev., 2022, 122, 8261–8312 CrossRef CAS
- K. Kuciński and G. Hreczycho, Green Chem., 2020, 22, 5210–5224 RSC
- R. L. Melen, Chem. Soc. Rev., 2016, 45, 775–788 RSC
- D. Lewandowski, T. Cytlak, R. Kempe and G. Hreczycho, J. Catal., 2022, 413, 728–734 CrossRef CAS
- H. Stachowiak, K. Kuciński, F. Kallmeier, R. Kempe and G. Hreczycho, Chem. – Eur. J., 2022, 28, e202103629 CrossRef CAS PubMed
- K. Kuciński and G. Hreczycho, ChemCatChem, 2022, e202200794, DOI:10.1002/cctc.202200794
- K. Kuciński, H. Stachowiak and G. Hreczycho, Eur. J. Org. Chem., 2020, 4042–4049 CrossRef
- E. O. Pentsak, D. B. Eremin, E. G. Gordeev and V. P. Ananikov, ACS Catal., 2019, 9, 3070–3081 CrossRef CAS
- S. Sau, M. Pramanik, A. Bal and P. Mal, Chem. Rec., 2022, 22, e202100208 CrossRef CAS PubMed
- X. Zhang, J. Fang, C. Cai and G. Lu, Chin. Chem. Lett., 2021, 32, 1280–1292 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data including NMR spectra. See DOI: https://doi.org/10.1039/d2cc04413g |
| This journal is © The Royal Society of Chemistry 2022 |