
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystalline phosphino(silyl)carbenes that readily form transition metal complexes†‡
Pawel
Löwe
 a and
Fabian
Dielmann
*b
a and
Fabian
Dielmann
*b
aInstitut für Anorganische und Analytische Chemie, Westfälische Wilhelms-Universität Münster, Corrensstraße 30, 48149 Münster, Germany
bInstitute of General, Inorganic and Theoretical Chemistry, Leopold-Franzens-Universität Innsbruck, Innrain 80-82, 6020 Innsbruck, Austria. E-mail: Fabian.Dielmann@uibk.ac.at
First published on 28th September 2022
Abstract
Phosphino(silyl)carbenes are known for their ineptitude to form transition metal complexes. We describe the synthesis of phosphino(silyl)carbenes bearing N-heterocyclic imine groups and show that these isolable, crystalline carbenes readily form stable copper(I) and gold(I) complexes. The solid-state structures of the free carbenes and their transition metal complexes are reported.
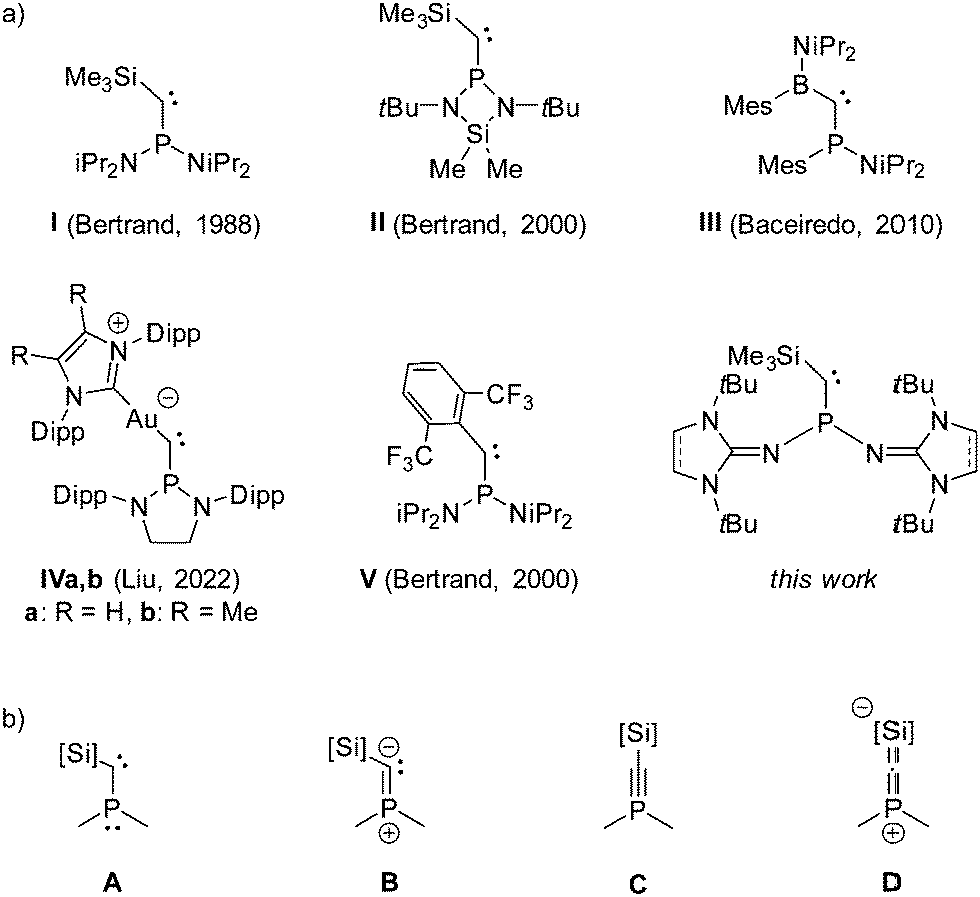
Since the first isolation of stable carbenes by Bertrand and co-workers in 1988 (Fig. 1a, I),1,2 and by Arduengo and co-workers in 1991 (1,3-diadamantylimidazolin-2-ylidene),3 the class of isolable singlet carbenes vastly expanded, covering a wide range of substitution patterns.4–6 Great efforts have been made to understand how their reactivity and donor properties can be tailored, particularly for the use as ligands in transition metal catalysis.5,7 The stability of phosphino(silyl)carbenes (e.g.I and II) is attributed to the mesomeric push-pull effect of the substituents at the carbene carbon atom. Due to the efficient π-donation from the phosphino group to the carbene carbon atom, phosphino(silyl)carbenes are best described as vinyl ylides (Fig. 1b, B) with modest contributions from the phosphaacetylene (C) and heterocumulene (D) electronic structures.8C and D represent the negative hyperconjugation of the carbene lone pair into σ* orbitals centered at phosphorus and silicon, leading to a widening of the carbene bond angle. This structural hallmark results in significant energy requirement to form a bent carbene structure for metal complexation, which in turn leads to rather low metal–carbon bond energies.9 Therefore, apart from complexes with group 13 element chlorides ECl3 (E = Al, Ga, In),10 direct complexation of phosphino(silyl)carbenes to metals has not yet been reported.11
 | ||
| Fig. 1 (a) Selected examples of phosphinocarbenes and the phosphinocarbene presented in this work. Dipp = 2,6-diisopropylphenyl. (b) Possible electronic structures of phosphino(silyl)carbenes ([Si] = silyl group). | ||
One strategy towards more bent acyclic phosphinocarbenes and thus improved ligand properties was to replace the pull-type silyl group with a spectator-type alkyl or aryl substituent.12 Unfortunately, phosphino(alkyl)carbenes turned out to be unstable at ambient conditions owing to low barrier rearrangement reactions. The first formation of transition metal complexes with isolable phosphinocarbenes was reported by Bertrand and co-workers in 2002, who showed that the phosphino(aryl)carbene V binds to rhodium(I) complexes in either η1 or η2 coordination mode along the P–C bond.13,14 Yet, phosphinocarbene complexes remain rare, although persistent phosphinocarbenes with various substitution patterns have been prepared,6 including the more recent phosphino(boryl)carbene III and the phosphino(auro)carbene IV.15,16
We herein report an alternative approach to form more stable phosphinocarbene complexes by modifying the phosphino group with N-heterocyclic imine (NHI) substituents. NHI groups are significantly stronger π donors than amino groups and have found broad application for the stabilization of low-valent main group and metallic species.17 We previously showed that NHI groups are extremely effective in increasing the electron donor ability of phosphines.18 From these results, we reasoned that NHI-substituted phosphino(silyl)carbenes should exhibit enhanced π-donation from the phosphino group to the carbene center and reduced back-bonding of the carbene lone pair, resulting in an overall increased vinyl ylide character (B).
Phosphino(silyl)carbenes 3a,b were prepared based on the established route: Reaction of the phosphenium chloride salts 1a,b19 with lithio-(trimethylsilyl)diazomethane gave the diazomethane derivatives 2a,b, which were used directly for the next step. Irradiation of the orange solutions of 2a,b in n-hexane with UV light at 365 nm or with sunlight led to the formation of carbenes 3a,b as yellow crystalline material (Scheme 1).
 | ||
| Scheme 1 Synthesis of phosphino(silyl)carbenes 3a,b. | ||
Phosphino(silyl)carbenes 3a,b are stable at ambient conditions.§ The 31P NMR signals of 3a,b (3a: −7.3 ppm, 3b: 19.9 ppm) are deshielded compared to phosphinocarbenes I (−40 ppm),1II (−26.7 ppm),20III (−36.7 ppm),15IVa,b (a: −17.1 ppm, b: −20.7 ppm),16 and V (−22.2 ppm).13 The 13C NMR resonance of the carbene carbon atom (3a: 75.1 ppm, 3b: 77.4 ppm) is shifted to lower frequencies compared to I (142.8 ppm),1 but is in the range of II (77.6 ppm).20 An exceptional feature of 3a,b are the small 1JPC coupling constants (3a: 49 Hz, 3b: 47 Hz) compared to the other phosphinocarbenes I (159 Hz),1II (153 Hz)20III (209 Hz),15IVa,b (a: 79 Hz, b: 77 Hz)16 and V (271 Hz).13 A similar trend has been observed for amino(phosphino)carbenes of type R2PCN(iPr)2, showing that the 1JPC coupling constants are significantly smaller for R = N(iPr)2 (23 Hz) than for R = Ph (102 Hz).21
Crystals obtained in the synthesis of 3a,b were suitable for X-ray diffraction (XRD) studies (Fig. 2). The solid-state structures of 3a and 3b reveal planar central SiCPN2 units flanked by the bulky tert-butyl groups of the NHI substituents. The P atoms in both compounds are in perfectly planar environments (sums of angles: 360°). The P–C bond lengths (3a: 1.582 Å, 3b: 1.589 Å) are elongated compared to those of II (1.532 Å)20 and V (1.544 Å),13 consistent with a reduced contribution of electronic structure C, however, they are shorter than the P–C bond in methylenephosphonium ion (iPr2N)2P = C(SiMe3)2+ (1.620 Å).22
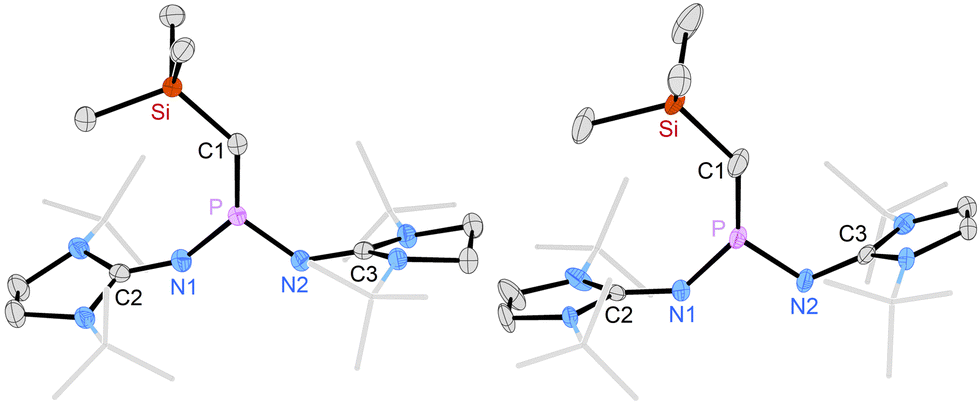 | ||
| Fig. 2 Solid-state structures of 3a (left) and 3b (right). Hydrogen atoms are omitted and tert-butyl groups are shown in wireframe for clarity. Ellipsoids are drawn at 50% probability. Selected bond lengths [Å] and angles [°]: 3a: C1–Si 1.786(2), C1–P 1.582(2), P–N1 1.596(2), P–N2 1.593(2), Si–C1–P 131.75(12), C2–N1–P 147.9(2), C3–N2–P 133.18(14). 3b: C1–Si 1.781(2), C1–P 1.589(2), P–N1 1.594(2), P–N2 1.6146(14), Si–C1–P 134.84(12), C2–N1–P 138.87(13), C3–N2–P 121.43(12). | ||
Most strikingly, the carbene bond angles in 3a (132°) and 3b (135°) are significantly smaller than those of phosphinocarbenes II (153°),20IVb (145°),16V (162°),13 and (iPr2N)2P–C-Mes (149°, Mes = 2,4,6-trimethylphenyl).12 With respect to their suitability as ligands in transition metal complexes, the bent structure of phosphino(silyl)carbenes 3a,b is beneficial because complexation is associated with less conformational energy penalty.9 Preliminary DFT calculations on 3a,b and the model phosphino(silyl)carbene (Me2N)2PCSiMe3 (3c) show that substitution of the amino groups with the NHI substituents leads to higher energies of the HOMO which corresponds to the carbene lone pair (3a: −4.216 eV, 3b: −4.035 eV, 3c: −5.143 eV) and of the π*-type acceptor orbitals (3a: 1.012 eV, 3b: 1.077 eV, 3c: 0.222 eV), indicating that 3a,b are better σ donors and weaker π acceptors than 3c (for details see ESI‡).
The new carbene 3a readily reacts with AuCl(tht) (tht = tetrahydrothiophene) to give complex 4 as a colorless solid in 97% yield (Scheme 2a). Treatment of carbene 3b with one equivalent of CuOtBu in THF or benzene resulted in the formation of a white solid that is insoluble in common solvents. We suspect that complex 5 forms an intermolecular coordination polymer in the solid state (Scheme 2b), which was corroborated by the reaction of 5 with another equivalent of CuOtBu, leading to the bimetallic copper complex 6 in quantitative yield. Note that copper(I) alkoxide complexes of N-heterocyclic carbenes (NHCs) are well-established precursors for copper hydride complexes23 and are thus active catalysts for semihydrogenations,24,25 hydroalkoxylations,26 and hydroborations.24 The η1 coordination mode of the carbenes in 4 and 6 is indicated by their 31P NMR chemical shifts (4: 70.0 ppm, 6: 80.8 ppm), which appear in the range of methylenephosphonium salts27 and trigonal NHI-substituted phosphonium ions.28 Moreover, similar deshielding of the 31P NMR resonance upon η1 coordination has been observed for phosphino(aryl)carbene V in complex [Rh(V)(nbd)Cl] (100.8 ppm; nbd = norbornadiene).14 As expected, the 13C NMR signal of the carbene carbon atoms are shifted to lower frequencies upon complexation (4: 57.8 ppm, 6: 53.1 ppm). However, the 1JPC coupling constants (4: 89 Hz, 6: 57 Hz) are larger than those of the free carbenes, which is in contrast to complex [Rh(V)(nbd)Cl] (rhodium complex: 1JPC = 2 Hz, free V: 1JPC = 271 Hz).14
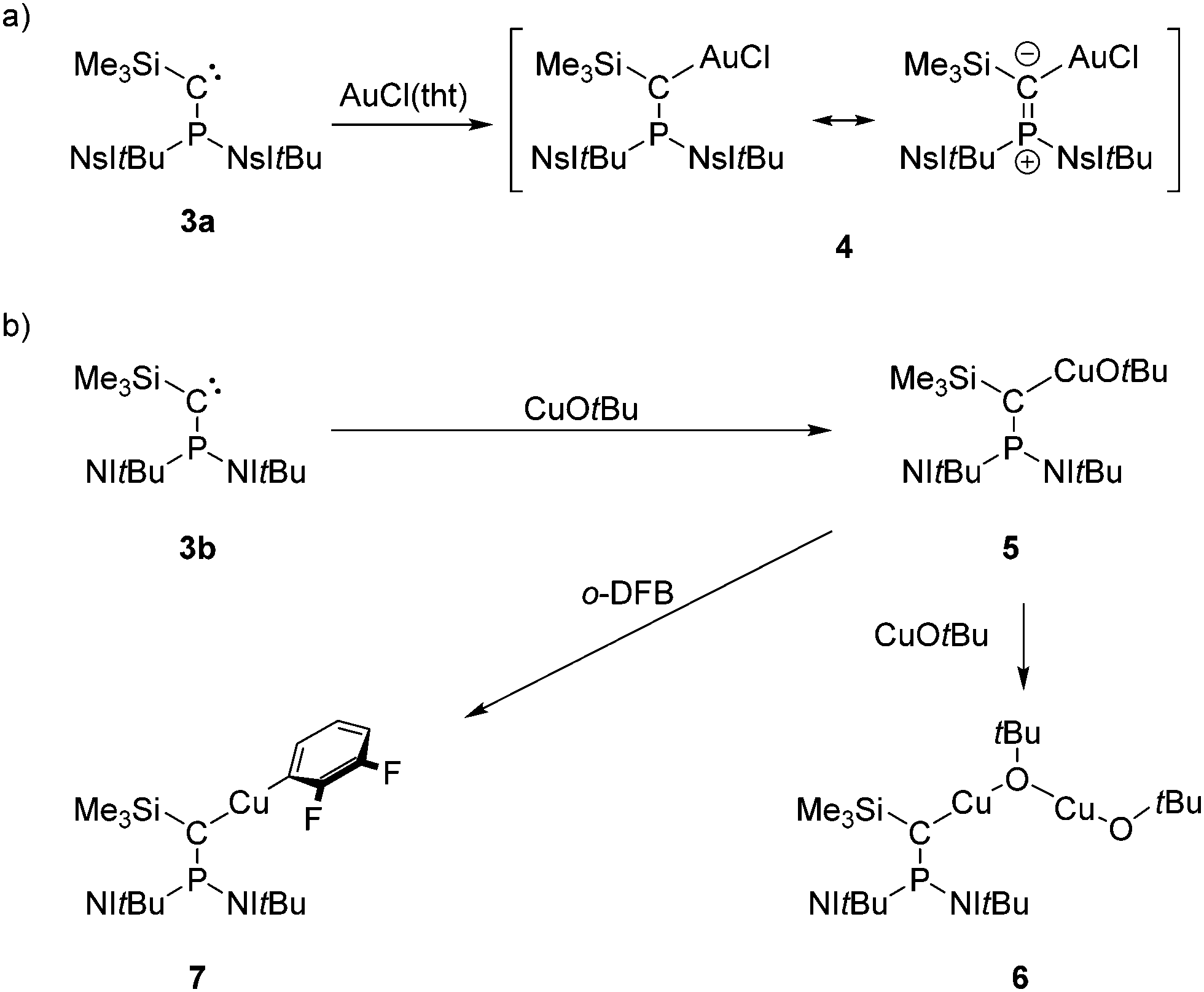 | ||
| Scheme 2 (a) Synthesis of gold(I) complex 4. (b) Synthesis of copper(I) complexes 5, 6 and 7. | ||
XRD quality crystals of 4 and 6 (Fig. 3, top) were obtained by cooling a saturated THF solution to −40 °C and by cooling a hot benzene solution to 21 °C, respectively. Due to the low solubility of 5 in THF, recrystallization was attempted in 1,2-difluorobenzene, leading to the formation of few crystals of complex 7, which were suitable for an XRD study (Fig. 3, bottom). Complex 7 presumably results from a C–H activation of 1,2-difluorobenzene at complex 5 but could not be characterized spectroscopically due to lack of substance.
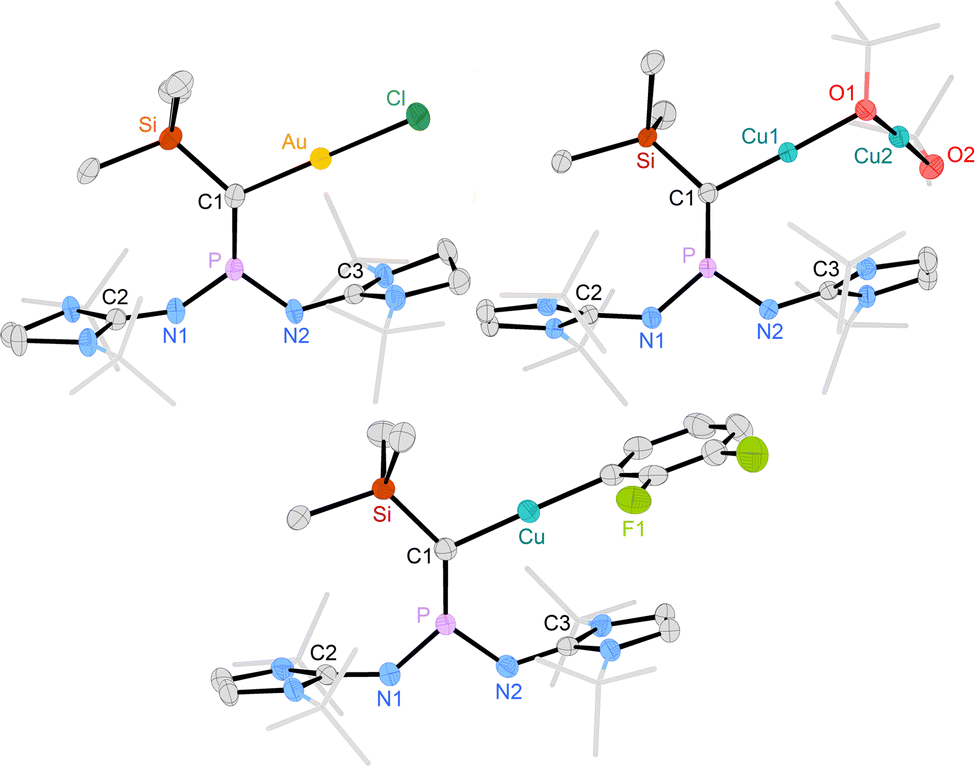 | ||
| Fig. 3 Solid-state structures of 4 (top left), 6 (top right) and 7 (bottom). Hydrogen atoms are omitted and tert-butyl groups are shown in wireframe for clarity. Ellipsoids are drawn at 50% probability. | ||
Selected geometrical parameters of 4, 6 and 7 are summarized in Table 1. The carbene–metal bond distances are comparable to those of NHC complexes (e.g. (ItBu)AuCl: 2.018 Å29 and (ItBu)CuOtBu: 1.876 Å;30 ItBu = 1,3-di-tert-butylimidazolin-2-yliden).31 In 4, 6 and 7 as well as the free carbenes 3a,b the P–N1–C2 angles are larger than the P–N2–C3 angles, which can be explained by the high steric demand of the SiMe3 group. The phosphorus–carbon bonds are slightly elongated in 4, 6 and 7 compared to those of the free carbenes (3a: 1.582 Å, 3b: 1.589 Å). In the metal complexes, both the carbene and the phosphorus atoms adopt a trigonal planar environment. It is noteworthy that all P–C–Si bond angles (4: 129.1°, 5: 133.8°, 7: 134.6°) are very similar to those of the free carbenes, suggesting that the conformational change of 3a,b upon metal complexation is indeed negligible. For comparison, the P–C–C bond angle of III (162.1°) is reduced by 43° in [Rh(III)(nbd)Cl] (119.0°).14
| Entry | 4 | 6 | 7 |
|---|---|---|---|
| C1–Si | 1.828(7) | 1.820(3) | 1.813(4) |
| C1–P | 1.624(7) | 1.610(3) | 1.605(5) |
| C1–M | 2.024(7) | 1.887(3) | 1.907(4) |
| Si–C1–M | 114.1(4) | 109.9(2) | 111.1(2) |
| M–C1–P | 116.1(4) | 116.1(2) | 113.8(2) |
| P–C1–Si | 129.1(4) | 133.8(2) | 134.6(3) |
| P–N1–C2 | 144.4(5) | 135.0(2) | 137.8(2) |
| P–N2–C3 | 128.7(5) | 125.9(2) | 126.3(3) |
| ∑C1(Si,M,P) | 359.3 | 359.8 | 359.5 |
| ∑P(C,N,N) | 360.0 | 360.0 | 360.0 |
Cavallo and co-workers established the % buried volume (% Vbur) model for the quantification of the steric impact of ancillary ligands.32 The value of 3a (50.1%) is higher than that of ItBu (39.3%)33 and sIDipp (1,3-bis(2,6-diisopropylphenyl)imidazolidine-2-ylidene) (47.4%),34 based on the geometry of the carbene-AuCl complexes. The calculated steric map (see ESI‡) indicates that the high value is mainly caused by the bulky NHI substituent flanking the metal atom. This structural feature explains why attempts to synthesize nonlinear complexes by reacting 3a with [(BiPr)PdBr2]2 (BiPr = 1,3-diisopropylbenzimidazolin-2-ylidene) or [(PEt3)PtCl2]2 were not successful.
In conclusion, the synthesis of two room-temperature stable, crystalline phosphino(silyl)carbenes 3a,b supported by bulky NHI substituents is reported. In contrast to other phosphinocarbenes, 3a,b directly form stable η1 complexes with CuI and AuI metal ions. Due to the strong π donor properties of the NHI groups, the free carbenes exhibit unusually small carbene bond angles which is advantageous for the formation of transition metal complexes. Thus, the use of NHI substituents could also improve the ligand properties of other carbenes.
The authors gratefully acknowledge financial support from the DFG (IRTG 2027). We thank Dr Tim Witteler and Dr Felix Kutter for their assistance in chemical synthesis, Dr Tristan T. Y. Tan for his help with the computational investigations and Dr Alexander Hepp for performing the NMR experiments and for his help with assigning the data.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463–6466 CrossRef CAS.
- A. Igau, A. Baceiredo, G. Trinquier and G. Bertrand, Angew. Chem., Int. Ed. Engl., 1989, 28, 621–622 CrossRef.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- (a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed; (b) H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS.
- J. Vignolle, X. Cattoën and D. Bourissou, Chem. Rev., 2009, 109, 3333–3384 CrossRef CAS PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS.
- (a) B. Römer, G. G. Gatev, M. Zhong and J. I. Brauman, J. Am. Chem. Soc., 1998, 120, 2919–2924 CrossRef; (b) D. A. Dixon, K. D. Dobbs, A. J. Arduengo and G. Bertrand, J. Am. Chem. Soc., 1991, 113, 8782–8785 CrossRef CAS; (c) N. Minh Tho, M. A. McGinn and A. F. Hegarty, Inorg. Chem., 1986, 25, 2185–2190 CrossRef CAS; (d) L. Nyulászi, D. Szieberth, J. Réffy and T. Veszprémi, THEOCHEM, 1998, 453, 91–95 CrossRef.
- W. W. Schoeller, D. Eisner, S. Grigoleit, A. B. Rozhenko and A. Alijah, J. Am. Chem. Soc., 2000, 122, 10115–10120 CrossRef CAS.
- A. H. Cowley, F. P. Gabbaï, C. J. Carrano, L. M. Mokry, M. R. Bond and G. Bertrand, Angew. Chem., Int. Ed. Engl., 1994, 33, 578–580 CrossRef.
- Note that phosphino(silyl)carbene complexes of scandium and uranium were prepared by generating Ph2P-C-SiMe3 via ligand modification in the coordination sphere of the metal atoms: (a) E. Lu, J. T. Boronski, M. Gregson, A. J. Wooles and S. T. Liddle, Angew. Chem., Int. Ed., 2018, 57, 5506–5511 CrossRef CAS PubMed; (b) W. Mao, L. Xiang, C. Alvarez Lamsfus, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2017, 139, 1081–1084 CrossRef CAS PubMed.
- E. Despagnet, H. Gornitzka, A. B. Rozhenko, W. W. Schoeller, D. Bourissou and G. Bertrand, Angew. Chem., Int. Ed., 2002, 41, 2835–2837 CrossRef CAS.
- C. Buron, H. Gornitzka, V. Romanenko and G. Bertrand, Science, 2000, 288, 834–836 CrossRef CAS.
- E. Despagnet, K. Miqueu, H. Gornitzka, P. W. Dyer, D. Bourissou and G. Bertrand, J. Am. Chem. Soc., 2002, 124, 11834–11835 CrossRef CAS PubMed.
- F. Lavigne, E. Maerten, G. Alcaraz, N. Saffon-Merceron, C. Acosta-Silva, V. Branchadell and A. Baceiredo, J. Am. Chem. Soc., 2010, 132, 8864–8865 CrossRef CAS PubMed.
- C. Hu, X.-F. Wang, R. Wei, C. Hu, D. A. Ruiz, X.-Y. Chang and L. L. Liu, Chem, 2022, 8(8), 2066–2068 Search PubMed.
- (a) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed; (b) N. Kuhn, M. Göhner, M. Grathwohl, J. Wiethoff, G. Frenking and Y. Chen, Z. Anorg. Allg. Chem., 2003, 629, 793–802 CrossRef CAS; (c) T. Ochiai, D. Franz and S. Inoue, Chem. Soc. Rev., 2016, 45, 6327–6344 RSC; (d) X. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116–138 CrossRef CAS.
- (a) M. A. Wünsche, P. Mehlmann, T. Witteler, F. Buß, P. Rathmann and F. Dielmann, Angew. Chem., Int. Ed., 2015, 54, 11857–11860 CrossRef PubMed; (b) P. Mehlmann, C. Mück-Lichtenfeld, T. T. Y. Tan and F. Dielmann, Chem. – Eur. J., 2017, 23, 5929–5933 CrossRef CAS PubMed; (c) J. A. Werra, M. A. Wünsche, P. Rathmann, P. Mehlmann, P. Löwe and F. Dielmann, Z. Anorg. Allg. Chem., 2020, 646, 794–799 CrossRef CAS.
- M. D. Böhme, T. Eder, M. B. Röthel, P. D. Dutschke, L. F. B. Wilm, F. E. Hahn and F. Dielmann, Angew. Chem., Int. Ed., 2022, e202202190 Search PubMed.
- T. Kato, H. Gornitzka, A. Baceiredo, A. Savin and G. Bertrand, J. Am. Chem. Soc., 2000, 122, 998–999 CrossRef CAS.
- N. Merceron, K. Miqueu, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 2002, 124, 6806–6807 CrossRef CAS PubMed.
- A. Igau, A. Baceiredo, H. Gruetzmacher, H. Pritzkow and G. Bertrand, J. Am. Chem. Soc., 1989, 111, 6853–6854 CrossRef CAS.
- (a) A. J. Jordan, G. Lalic and J. P. Sadighi, Chem. Rev., 2016, 116, 8318–8372 CrossRef CAS PubMed; (b) Homogeneous hydrogenation with non-precious catalysts, ed. J. F. Teichert, Wiley-VCH, Weinheim, Germany, 2020 Search PubMed.
- J. W. Hall, D. M. L. Unson, P. Brunel, L. R. Collins, M. K. Cybulski, M. F. Mahon and M. K. Whittlesey, Organometallics, 2018, 37, 3102–3110 CrossRef CAS.
- (a) K. Semba, T. Fujihara, T. Xu, J. Terao and Y. Tsuji, Adv. Synth. Catal., 2012, 354, 1542–1550 CrossRef CAS; (b) A. M. Whittaker and G. Lalic, Org. Lett., 2013, 15, 1112–1115 CrossRef CAS; (c) G.-H. Wang, H.-Y. Bin, M. Sun, S.-W. Chen, J.-H. Liu and C.-M. Zhong, Tetrahedron, 2014, 70, 2175–2179 CrossRef CAS; (d) F. Pape, N. O. Thiel and J. F. Teichert, Chem. – Eur. J., 2015, 21, 15934–15938 CrossRef CAS PubMed.
- C. Munro-Leighton, S. A. Delp, E. D. Blue and T. B. Gunnoe, Organometallics, 2007, 26, 1483–1493 CrossRef CAS.
- O. Guerret and G. Bertrand, Acc. Chem. Res., 1997, 30, 486–493 CrossRef CAS.
- (a) F. Dielmann, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 14071–14073 CrossRef CAS PubMed; (b) M. A. Wünsche, T. Witteler and F. Dielmann, Angew. Chem., Int. Ed., 2018, 57, 7234–7239 CrossRef PubMed; (c) P. Löwe, T. Witteler and F. Dielmann, Chem. Commun., 2021, 57, 5043–5046 RSC; (d) P. Mehlmann, T. Witteler, L. F. B. Wilm and F. Dielmann, Nat. Chem., 2019, 11, 1139–1143 CrossRef CAS PubMed.
- S. Singh, S. S. Kumar, V. Jancik, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, Eur. J. Inorg. Chem., 2005, 3057–3062 CrossRef CAS.
- J. Plotzitzka and C. Kleeberg, Inorg. Chem., 2016, 55, 4813–4823 CrossRef CAS PubMed.
- (a) P. de Frémont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411–2418 CrossRef; (b) T. Asada, Y. Hoshimoto and S. Ogoshi, J. Am. Chem. Soc., 2020, 142, 9772–9784 CrossRef CAS PubMed.
- (a) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed; (b) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- M. V. Baker, P. J. Barnard, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2005, 37–43 RSC.
- P. de Frémont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411–2418 CrossRef.
Footnotes |
| † Dedicated to Professor Guy Bertrand on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2190727–2190733. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04321a |
| § After storage of solid 3a,b under an argon atmosphere for more than one year, no decomposition was observed according to 1H and 31P NMR spectroscopy. |
| This journal is © The Royal Society of Chemistry 2022 |