
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical tools to track and perturb the expression of sialic acid and fucose monosaccharides
Emiel
Rossing
,
Johan F. A.
Pijnenborg
and
Thomas J.
Boltje
 *
*
Department of Synthetic Organic Chemistry, Institute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525AJ, Nijmegen, The Netherlands. E-mail: thomas.boltje@ru.nl
First published on 10th October 2022
Abstract
The biosynthesis of glycans is a highly conserved biological process and found in all domains of life. The expression of cell surface glycans is increasingly recognized as a target for therapeutic intervention given the role of glycans in major pathologies such as cancer and microbial infection. Herein, we summarize our contributions to the development of unnatural monosaccharide derivatives to infiltrate and alter the expression of both mammalian and bacterial glycans and their therapeutic application.
Introduction
Glycosylation, the expression of carbohydrate structures (glycans) on proteins and lipids, is found in all the domains of life. The collection of all glycans found on a cell is called the “glycome” which is information rich and a key player in a plethora of physiological and pathological processes.1 The information that the glycome holds can be written, read and erased by glycosyltransferases, lectins and glycosidases, respectively. Glycosyltransferase enzymes assemble glycans by the addition of monosaccharide building blocks. Due to variations in the monosaccharide type, order, glycosidic bond stereo- (α or β) and regiochemistry, glycans can be constructed with impressive structural complexity. Structural complexity is further driven by the fact that glycans can be long, short, branched or linear in structure and are often connected to proteins (N-glycans, O-glycans, proteoglycans), lipids (glycolipids) or both (glycosylphophatidylinositol-anchored proteins) thereby providing additional possibilities for their presentation and subsequent biological function.2Cell surface glycans are essential mediators of biological processes. For example, cell surface glycans can be recognized by receptors expressed on immune cells to enable the detection of host and pathogen derived glycans to attenuate or activate the immune system, respectively. Furthermore, a growing body of literature demonstrates that the glycosylation pattern of cancer cells is different from that of healthy cells. It is still unclear whether aberrant glycosylation of cancer cells is a cause or consequence of tumorigenesis but it is associated with aggressive and invasive forms of cancer and hence poor prognosis.3
Due to their prominent role in various pathologies, glycans and their cognate receptors are emerging as new targets for drug discovery and development. Several distinct approaches targeting glycans for therapeutic effect are applied in the clinic or in (pre)clinical development (Fig. 1). For example, antibodies and vaccine strategies directed at unique bacterial glycans or tumor associated carbohydrate antigens (TACAs) enable the host to elicit a potent targeted immune response (Fig. 1A). Immune receptors binding glycans can be targeted using antibodies or small molecules to break receptor-glycan interactions (Fig. 1B).4,5 Alternatively, the glycan ligand can be degraded using glycosidase enzymes to disrupt receptor binding (Fig. 1C).6 Small molecule inhibitors of glycan binding receptors can also be used to block their binding, such as Uproleselan targeting selectins or zanamivir targeting pathogen sialidases (Fig. 1D and E).
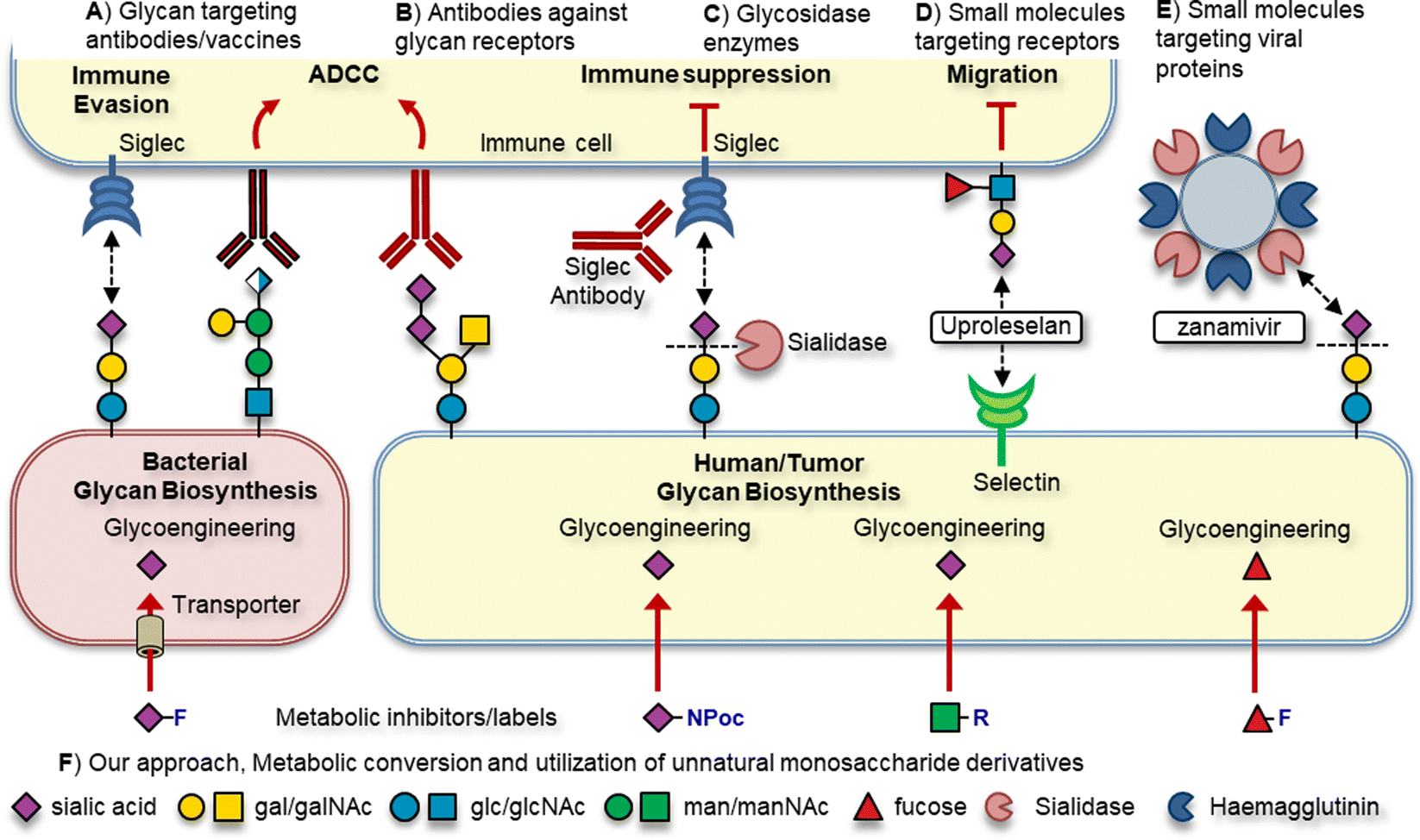 | ||
| Fig. 1 Overview of the therapeutic strategies targeting glycosylation. | ||
Another strategy targets the intracellular biosynthesis pathways that are responsible for the construction of glycoconjugates using metabolic oligosaccharide engineering (MOE, Fig. 1F). MOE has several advantages over the aforementioned strategies. For example, targeting the intracellular biosynthesis allows for the inhibition or labelling of extracellular and intracellular forms of glycosylation. Additionally, exploiting differences in monosaccharide uptake and biosynthesis pathways between host and pathogen allows for the selective targeting of specific forms of glycosylation.7 The basic principle of MOE is to feed cells chemically modified monosaccharide derivatives that can be metabolically processed by the enzymes involved in glycoconjugate assembly. To enable their cellular uptake, derivatives need to be recognized by their respective monosaccharide transporters. Alternatively, monosaccharides that are not actively transported cells can be administered in peracetylated form to increase their lipophilicity enabling passive cellular uptake. Inside cells, the ester groups are cleaved by esterase enzymes affording the unnatural monosaccharide derivative which is then metabolically processed.7 As the enzymes involved in metabolic processing are rather specific, only small alterations to the natural monosaccharide are tolerated. By the introduction of small modifications on monosaccharides designed to block a specific enzymatic step, the glycosylation pathway can be inhibited. Small modifications such as deoxygenation/fluorination of monosaccharides are typically used to maintain metabolic processing towards the active metabolite, the corresponding nucleotide sugar.
MOE can also be used to enable the metabolic labelling of glycoconjugates to enable their detection and isolation. To this end, monosaccharides carrying a small chemical reporter group such as an azide or alkyne can be incorporated into glycans leading to glycoconjugates carrying these chemical reporters. Using bioorthogonal chemistry, the chemical reporter group can be reacted with its cognate reaction partner to introduce functional handles that allow for the detection, isolation and analysis of the labelled glycoconjugate.8 MOE strategies designed to label and/or inhibit mammalian glycosylation can also be used to target homologous pathways in other organisms such as bacteria. By exploiting differences in monosaccharide transporter expression and ensuing glycosylation pathways, the specific targeting of bacteria vs host glycosylation can be achieved.9–11
Herein we will review our recent contributions to the development of metabolic probes to either inhibit or label specific monosaccharides and their applications. A particular focus will be on tools to modulate the expression of sialic acids and fucose monosaccharides as they are important capping monosaccharides. For a more broad overview of glycomimetics targeting other classes of monosaccharides, such as O-GlcNAcylation we refer to a recent review by Vocadlo and co-workers.12
Metabolic inhibitors of mammalian fucosylation
Glycans or proteins are fucosylated by the action of thirteen fucosyltransferases (FTs) that differ in acceptor glycan preference but all utilize GDP-fucose as the donor substrate. Cells biosynthesize GDP-fucose either via the recycling of fucose released during glycan turnover in the lysosome (salvage pathway) or via the de novo biosynthesis from mannose-1-phosphate. Fluorinated sugars derived from the nucleotide sugar donors have been shown to be potent inhibitors of glycosyltranferases. For example, Wong and co-workers demonstrated that fucosyltransferases can be inhibited by GDP-2-deoxy-2-fluorofucose which acts as a competitive inhibitor.13 However, this compound is very polar and cannot traverse the cell membrane and hence cannot be used to inhibit fucosylation in cells. This hurdle was overcome by Paulson and co-workers who employed 1,3,4-tri-O-acetyl-2-deoxy-2-fluorofucose (2FF, 1), a cell permeable metabolic precursor of GDP-2-deoxy-2-fluorofucose (Fig. 2). 2FF is passively taken up by cells and deacetylated by intracellular esterases to afford 2-deoxy-2-fluorofucose which is metabolized by fucose kinase and GDP-L-fucose pyrophosphorylase (GFPP) to form the bioactive GDP-analog. GDP-2-deoxy-2-fluoro-fucose competitively binds FTs and blocks their activity. Moreover, GDP-2-deoxy-2-fluorofucose induces feedback inhibition of the de novo biosynthesis by the allosteric inactivation of GMDS.14 Combined, these effects ensure suppression of fucosylation in cells and 2FF has been extensively used to investigate the role of fucosylation in various pathologies.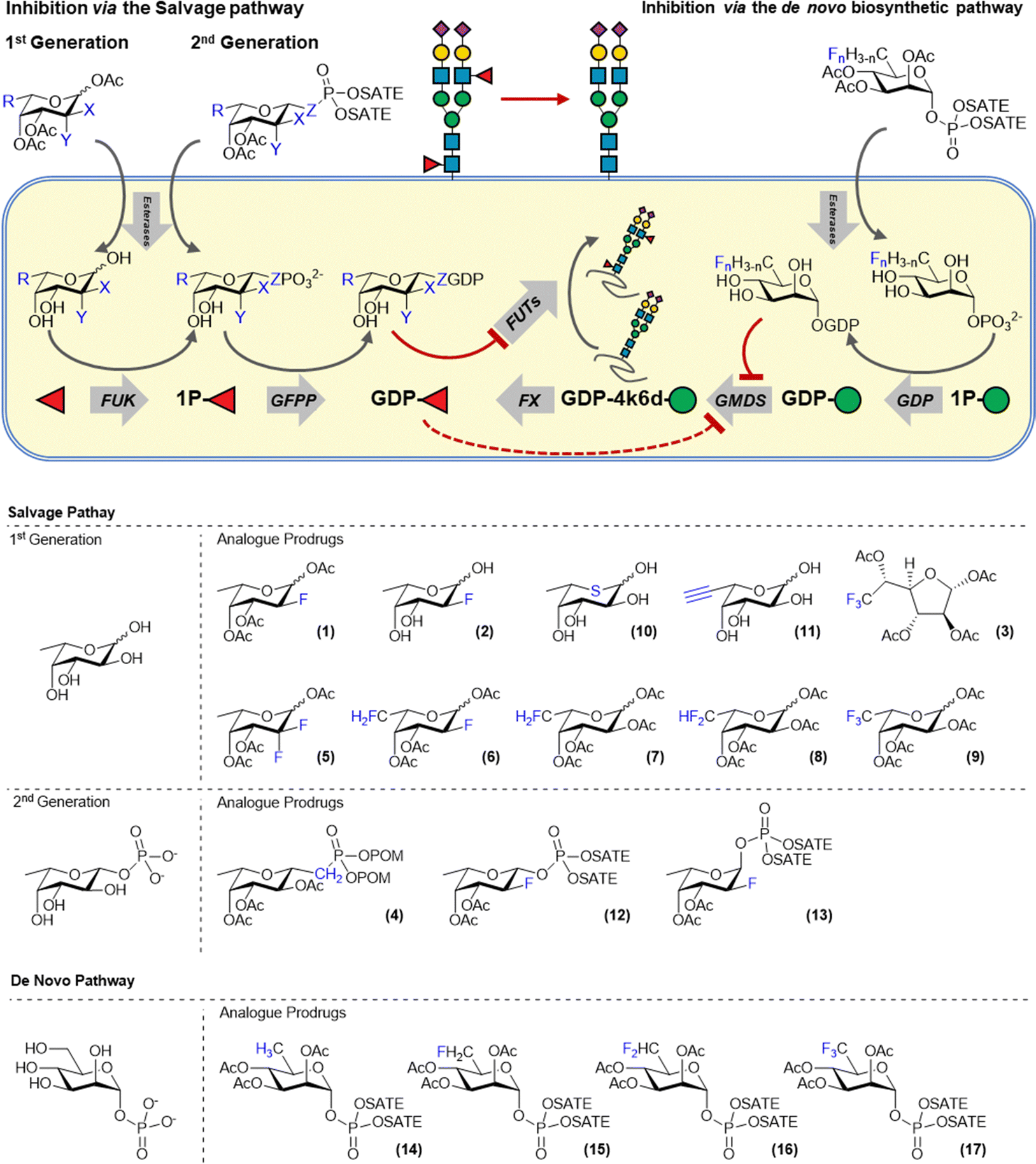 | ||
| Fig. 2 Overview of the fucose biosynthesis and inhibitors targeting the salvage and de novo biosynthesis pathway. | ||
For example, application of 2FF, or its deacetylated form, investigating the effect of tumor fucosylation in liver and breast cancer cells resulted in reduced proliferation, migration, and downstream growth factor activation.15,16In vivo studies showed that pre-treatment of HepG2 liver cancer cells reduced outgrowth after subcutaneous transplantation in mice.15 Oral administration of 2FF delayed tumor growth of LS174T colorectal carcinoma, breast cancer and A20 lymphoma in mice, likely due to enhanced ADCC after fucose inhibition.17–19 Oral 2FF treatment combined with immunotherapy completely protected against tumor growth due to enhanced ADCC in A20 lymphoma cells, with no apparent toxicity in mice.17 Very recently, a Phase I human clinical trial was performed with 2FF, making it the first in-human use of this type of metabolic inhibitor. Patients with advanced solid tumors were given daily oral doses of 2–15 g of 2FF in a dose range finding study. A significant drop in tumor burden was found, however the clinical trial was terminated following thromboembolic events.20
The efficacy of 2-deoxy-2-fluorofucose has inspired the development of a number of additional metabolic fucosylation inhibitors (3–13, Fig. 2). Most of these are also based on L-fucose and utilize the salvage pathway to afford a bioactive GDP-fucose derivative inside cells. For example, McCarter and co-workers developed a range of modified fucose derivatives and tested them on antibody producing hybridoma cells. Fucostatin I, which is a 6,6,6-trifluorofucose derivative (3) was found to inhibit core-fucosylation more efficiently than 2FF (1).21 However, residual incorporation of both unnatural fluorosugars into the antibody glycans was observed. Therefore, Fucostatin II (4) was developed, a fucose-1-phosphonate analog. The anomeric phosphonate group allows for the metabolic processing towards the GDP-derivative but cannot be transferred by fucosyltransferases. This mode of action prevents metabolic incorporation of the unnatural derivative into glycans.21 Wang and co-workers systematically investigated the effect of fucose fluorination by the preparation of C-2 and C-6 mono-di- and trifluorinated fucose derivatives (5–9). The effect of this panel on cancer cell proliferation was investigated and the largest effects were observed for 6,6-difluorofucose (8) and 6,6,6-trifluorofucose (9). Inhibition of FUT-8 by the GDP-analogs of these compounds was also investigated with the best inhibition observed for GDP-2FF.22 Another fucose based metabolic inhibitor operates via the substitution of the endocyclic oxygen for sulphur. As described earlier for a 5-thio-GlcNAc derivative, the corresponding 5-thio-fucose derivative (10) acts as an inhibitor and was used to block antibody fucosylation.23 Core-fucosylation was inhibited by up to 95% in Chinese Hamster Ovary cells. However, 5-thio-L-fucose was also incorporated onto glycans up to 50% of the physiological fucosylation levels of cellular glycans.24 6-alkynyl-fucose (11), originally developed to metabolically label fucose residues, was also shown to exhibit inhibitory properties. Kizuka et al. showed that 6-alkynyl-fucose inhibited fucosylation in liver cancer cells and inhibited cellular migration and tissue-invasion. The mechanism of inhibition was elucidated and it was demonstrated that the active metabolite GDP-6-alkynylfucose inhibited the de novo biosynthesis of GDP-fucose by inhibiting the FX enzyme but did not have any activity on GMDS.25 Besides its activity as a metabolic inhibitor, 6-alkynyl-fucose (11) is also partially incorporated into glycoproteins by fucosyltransferases.26
Our lab has also contributed to the development of novel fucosylation inhibitors. We reasoned that the metabolic conversion of 2FF (2) towards its bioactive GDP-derivative inside cells could be improved by feeding cells a cell-permeable 2-deoxy-2-fluorofucose-1-phosphate derivative. Feeding cells with a fucose-1-phosphate derivative bypasses the action fucose kinase and hence requires only the action of GFPP to afford the bioactive GDP-derivative. To enable the passive uptake of 2-deoxy-2-fluorofucose-1-phosphate derivatives, S-acetyl-2-thioethyl (SATE) groups were used to cage the anomeric phosphate group.27 During the chemical synthesis, both the alpha (A2FF1P, 13) and beta (B2FF1P, 12) anomeric phosphate derivatives were formed and both were tested on cells. The natural beta anomer (B2FF1P, 12) was indeed more potent than 2FF (1) which we attribute to the more efficient metabolic processing to its bioactive GDP-derivative as B2FF1P (12) bypasses the fucose kinase step. This is supported by the analysis of the intracellular nucleotide sugar pool which showed an increased level of GDP-2-deoxy-2-fluorofucose when B2FF1P (12) was used compared to 2FF. To our surprise, the unnatural alpha anomer (A2FF1P, 13) also acted as an inhibitor of fucosylation. Intracellular nucleotide sugar analysis demonstrated that this derivative is also converted to its α-GDP-2-deoxy-2-fluorofucose derivative. Fluorination was critical for bioactivity as α-fucose-1-phosphate did not act as an inhibitor of fucosylation.
All metabolic fucosylation inhibitors mentioned above utilize the fucose salvage pathway to achieve the inhibition of cellular fucosylation. Recently we reported the design, synthesis and biological evaluation of inhibitors that target the de novo fucose biosynthesis. It is estimated that, depending on the cell-type, around ∼90% of the GDP-fucose pool is biosynthesized via the de novo biosynthesis from GDP-mannose.28 We reasoned that directly targeting GMDS, an enzyme critical to the biosynthesis of fucose, might yield more potent fucosylation inhibitors. GMDS is a 4,6-dehydratase enzyme that converts GDP-mannose to GDP-4-keto-6-deoxy-mannose which is subsequently processed by FX, a 3,5-epimerase-4-ketoreductase, to afford GDP-fucose. We were inspired by Liu and co-workers, who showed that CDP-glucose-4,6-dehydrate from Yersinia pseudotuberculosis, a bacterial homolog of GMDS, could be inhibited by the C-5 difluoromethyl sugar derivative of its monosaccharide substrate (CDP-glucose).29 Hence, we designed and synthesized C-5 mono-, di- and trifluororhamnose-1-phosphate derivatives (14–17) to investigate their metabolic processing and ability to inhibit GMDS. We expected these derivatives to be passively taken-up, deacylated and metabolically processed by GDP to afford the corresponding bioactive fluorinated-GDP-rhamnosides inside cells. Indeed, intracellular nucleotide sugar analysis showed the formation of the corresponding fluorinated GDP-rhamnosides derivatives inside cells and inhibition of fucosylation of the di- and trifluoro derivatives FucoTrim I and II, respectively, was confirmed by a loss of lectin binding. Subsequent mechanistic analysis on recombinant GMDS showed that FucoTrim I mainly acts as a competitive inhibitor for GMDS.30
The ever expanding toolbox of metabolic inhibitors of fucosylation enables the investigation of their therapeutic potential, mainly in the context of cancer development and progression.
Metabolic inhibitors of mammalian sialylation
Sialoglycans are biosynthesized via the de novo biosynthesis of sialic acid which derived from the metabolism of glucose. N-Acetylglucosamine (UDP-GlcNAc), is converted by the bifunctional enzyme UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE) into N-acetylmannosamine (ManNAc) via its epimerase domain (GNE) and subsequently produces ManNAc-6-phosphate using its kinase activity (MNK). ManNAc-6-phosphate is converted into Neu5Ac-9-phosphate by the N-acetylneuraminate synthase (NANS) and dephosphorylated by Neu5Ac-9-P-phosphatase (NANP) to yield Neu5Ac (Fig. 3). Another source of Neu5Ac is provided by sialidase mediated breakdown of sialoglycans in the lysosome. Released Neu5Ac can be transported into the cytosol via the sialin (SLC17A5) transporter for recycling. In the nucleus, Neu5Ac is conjugated with cytidine monophosphate (CMP) by the CMP sialic acid synthase (CMAS) and transported into the Golgi system via the CMP–sialic acid transporter SLC35A1. Cytosolic CMP–sialic acid levels regulate the de novo synthesis of sialic acids by feedback inhibition of GNE. Twenty sialyltransferases (STs) utilize CMP-Sia but differ in acceptor glycan preference by created a complex array of sialoglycans.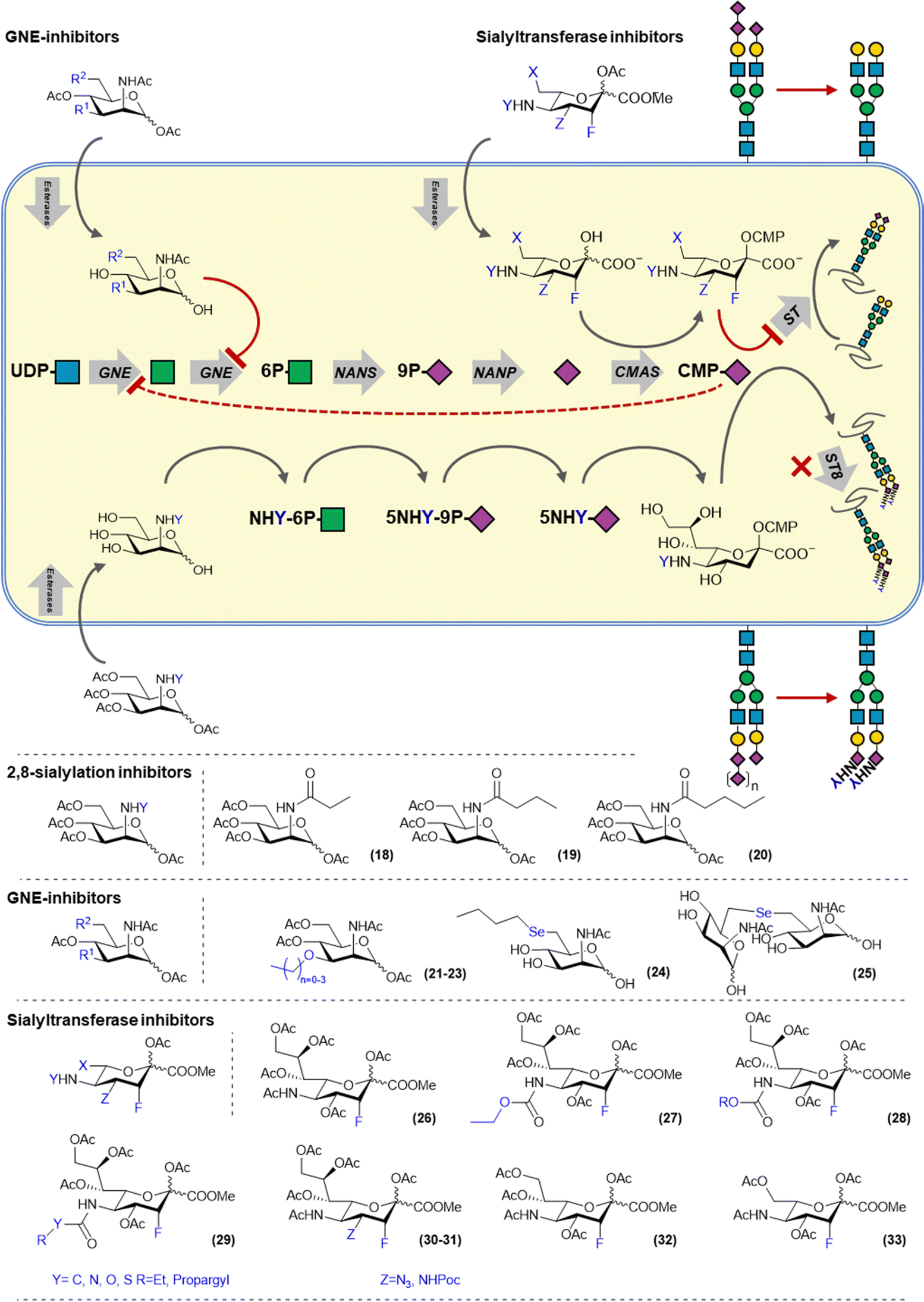 | ||
| Fig. 3 Overview of sialic acid biosynthesis and inhibitors targeting the salvage and de novo pathways. | ||
Metabolic inhibitors targeting the sialic acid biosynthesis pathway mainly target GNE and STs as the target enzymes. Reuter and co-workers have demonstrated that ManNAc derivatives carrying different N-acyl groups are tolerated by the de novo biosynthesis pathway and can be metabolized and incorporated into sialoglycans. However, shortly thereafter, Bertozzi and co-workers showed that introduction of propanoyl, butanoly- and pentanoyl amides onto mannosamine (ManNprop, ManNbut and ManNpent 18–20) led to a reduced expression of α-2,8 polysialic acids (PSA).31 This is due to the mannosamine derivatives being converted into their corresponding C-5 modified sialic acids, which elongated C-5 chain block the action of α-2,8-STs preventing the polymerisation of sialic acid to PSA (Fig. 3).32 MCF-7 breast cancer cells treated with these derivatives showed reduced migration and adhesion due to lower NCAM sialylation.33 Also other human carcinoma cell lines treated with ManNBut (18) showed reduced PSA on the NCAM carrier protein.31 Other ManNAc based sialylation inhibitors are based on C-3 modified derivative and reduce overall cellular sialylation by targeting the sialic acid de novo biosynthesis enzyme GNE. Reutter and co-workers showed that the ManNAc 3-O-methyl, -ethyl and n-propyl ethers (21–23) were able to inhibit sialylation, with the methyl ether (21) having an IC50 of 176 μM. They also showed that no sialic acid containing the modifications was detected in cell lysate, proving that the modification cannot be processed by GNE.34 Later, it was also shown that C-6 selenyl mannosamine derivatives (24–25) can also inhibit GNE by blocking the phosphorylation activity of the enzyme.35
Alternatively, sialic acid based inhibitors have been developed that enter the sialic acid biosynthesis via the recycling pathway (Fig. 3). Paulson and co-workers also developed a metabolic inhibitor of sialylation, based on 3-fluorosialic acid. The 3-fluorosialic acid was peracetylated (FNANA, 26) to facilitate its passive diffusion over the cell membrane. Once FNANA enters the cell, the ester groups are cleaved by esterases to yield the unprotected 3-fluorosialic acid. Conversion into its CMP-derivative by CMAS then afford to active inhibitor, 3-fluoro-CMP–sialic acid which competitively blocks the action of STs and induces feedback inhibition of GNE thereby also shutting down the de novo sialic acid biosynthesis.14
As the overexpression of sialoglycans in cancer is linked enhances tumor growth, FNANA has been used extensively to study tumor sialylation. FNANA was able to decrease sialylation efficiently in a B16F10 melanoma model which lead to decreased cell adhesion and migration, and a significant reduction in tumor growth and improved survival in vivo.36 More recently, significant anti-tumor activity was also demonstrated in an in vivo multiple myeloma model, as well as showing synergistic effects with CD38 targeting antibodies.37,38 Besides the effect of these untargeted approaches to desialylate tumors in vivo, FNANA has been formulated in melanoma-targeting nanoparticles enabling accumulation of FNANA inside the tumor, preventing metastasis of melanoma to the lungs.39 The biological mechanism by which tumor desialylation reduced its growth and metastasis was linked to immune mediated killing involving CD8+ T-cells as depletion of this immune cell subset abrogated the in vivo efficacy of FNANA as an antitumor compound. More recently, the underlying mechanism of immune mediated killing was elucidated. Büll et al. showed treating monocyte derived DCs with FNANA led to decreased binding of immunosuppressive Siglec receptors expressed on the DCs, enhancing the responsiveness of the DCs to Toll-like receptor (TLR) activation.40 Similarly, T-cell activation was shown to be dependent on co-stimulation of the TCR and CD28 receptors by DC antigen presenting MHC and CD80/86, respectively. Both cis- and trans-interactions between CD28 and cellular sialoglycans dampens this activation mechanism and could be enhanced by removal of sialic acids thereby increasing the avidity of the DC-T-cell interactions potentiating the immune response.41,42
To investigate the structure–activity relationship of FNANA and improve its inhibitory potency, we prepared and tested a variety of structural analogs. The C-5 position of sialic acid is amenable to modification as it has been previously shown that the sialic acid metabolism tolerates sialic acid and mannosamine analogs modified at this position.43 A small library of C-5-modified sialic acids were synthesized, containing several aliphatic and unsaturated side groups, linked to the C-5 amine through an amide or a carbamate. The potency of these new inhibitors was screened in different human and murine cancer cell-lines and interestingly, analogs bearing a carbamate (27–28) showed dramatically increased potency in vitro with some of the analogs showing inhibition in models that are insensitive to treatment with FNANA. Moreover, inhibition lasted considerably longer. Investigation of the mechanism of improved inhibitory potency of these derivatives revealed that they are more efficiently converted by CMAS to the corresponding CMP-derivative leading to higher intracellular concentrations of the bioactive inhibitor and hence a more potent and durable inhibition of the sialylation pathway.44
More recently, we explored modification at the C-4, C-5, C-8 and C-9 more extensively (29–33). The striking difference in activity between C-5 amides and carbamates sparked the interest in exploring different linkage types at that position, including the urea, thiourea and the S-thiocarbamates. All these linkages showed increased inhibition over the amide linkages, with the carbamates and thiocarbamates performing the best in vitro. Modifications at the glycerol tail were explored and it was discovered that the C-9 carbon can be removed without complete loss of inhibition or selectivity, making it a potential site for further modifications. Modifications at the C-4 (30–31) also still showed inhibition, yet not as potent as the C-5 modified 3-fluorosialic acids.45
Metabolic labelling of sialic acids to create selective Siglec ligands
The ability of unnatural monosaccharides analogs to navigate the metabolic pathways an incorporate into cell surface glycans has led to the development of bioorthogonal chemistry. Early work by Reuter and co-workers has demonstrated that mannosamine analogs carrying various unnatural amide analogs could be incorporated into sialoglycans. Inspired by this work, Bertozzi and co-workers sought to introduce a chemical reporter group in cell surface glycans by feeding unnatural mannosamine derivatives. The chemical reporter group is a small, non-toxic reactive functional group that can be chemo-selectively ligated under physiological conditions. After metabolic incorporation, the chemical reporter group is reacted with its cognate reaction partner under physiological conditions to enable the introduction of a fluorescent label or affinity tag to enable the detection and isolation of sialoglycans, respectively. This form of sialoglycan labelling has contributed to our understanding of sialoglycan expression and structure and has also served as an important proving ground for the development of new bioorthogonal reaction pairs such azide–phosphines, azide–alkynes, alkene–tetrazines. The principle of metabolic oligosaccharide engineering established for sialoglycans has now been extended to other mammalian and bacterial monosaccharides whilst bioorthogonal chemistry has been broadly adopted in molecular life science.7,8We have recently reported a combination of metabolic sialoglycan engineering and bioorthogonal chemistry to generate high affinity ligands targeting Siglec receptors. By generating synthetic ligands on living cells, improved immune suppression by increased Siglec binding was achieved. Siglecs are transmembrane receptors bearing an extracellular N-terminal V-set Ig (Ig–V) domain responsible for the binding of sialic acid ligands. Siglecs bind sialic acids on opposing (trans) or on the same cell (cis) generally resulting in immune-suppressive signalling. Siglecs bind sialic acids in a monovalent manner with low affinity (0.1–3 mM). It has been shown that chemically modified sialic acids can improve Siglec binding affinity and selectivity and that such modifications can be introduced via the copper-catalyzed alkyne–azide cycloaddition (CuAAC). To enable the multivalent presentation of these synthetic sialic acids, they are often incorporated into liposome carriers or on polymer scaffolds. Since sialoglycan engineering allows for the incorporation of alkyne or azide sialic acids and the CuAAC reaction is bioorthogonal, we investigated their combination to chemically modify sialic acids on living cells to enhance their binding to Siglecs whilst maintaining their natural multivalent presentation on the cell surface (Fig. 4). To this end, alkyne or azide modified sialic acids were metabolically incorporated into living cells and reacted with over sixty different azides/alkynes using CuAAC. The obtained library of glycoengineered cells, was screened for binding to the human Siglec family and led to the identification of modifications that dramatically increased Siglec binding (>100 fold). Using structure-guided design, sialic acid modifications highly selective for Siglec-5/-14 were developed. Finally, functional analysis showed that glycoengineered cells programmed to bind Siglec-3 were able to dampen the activation of Siglec-3+ immune cell. These high affinity Siglec ligand expressing cells (HASLECs) were able to significantly decrease activation of the NF-κB and IRF pathways in monocyte reporter cells expressing Siglec-3, when stimulated with LPS or Pam3CSK4.46
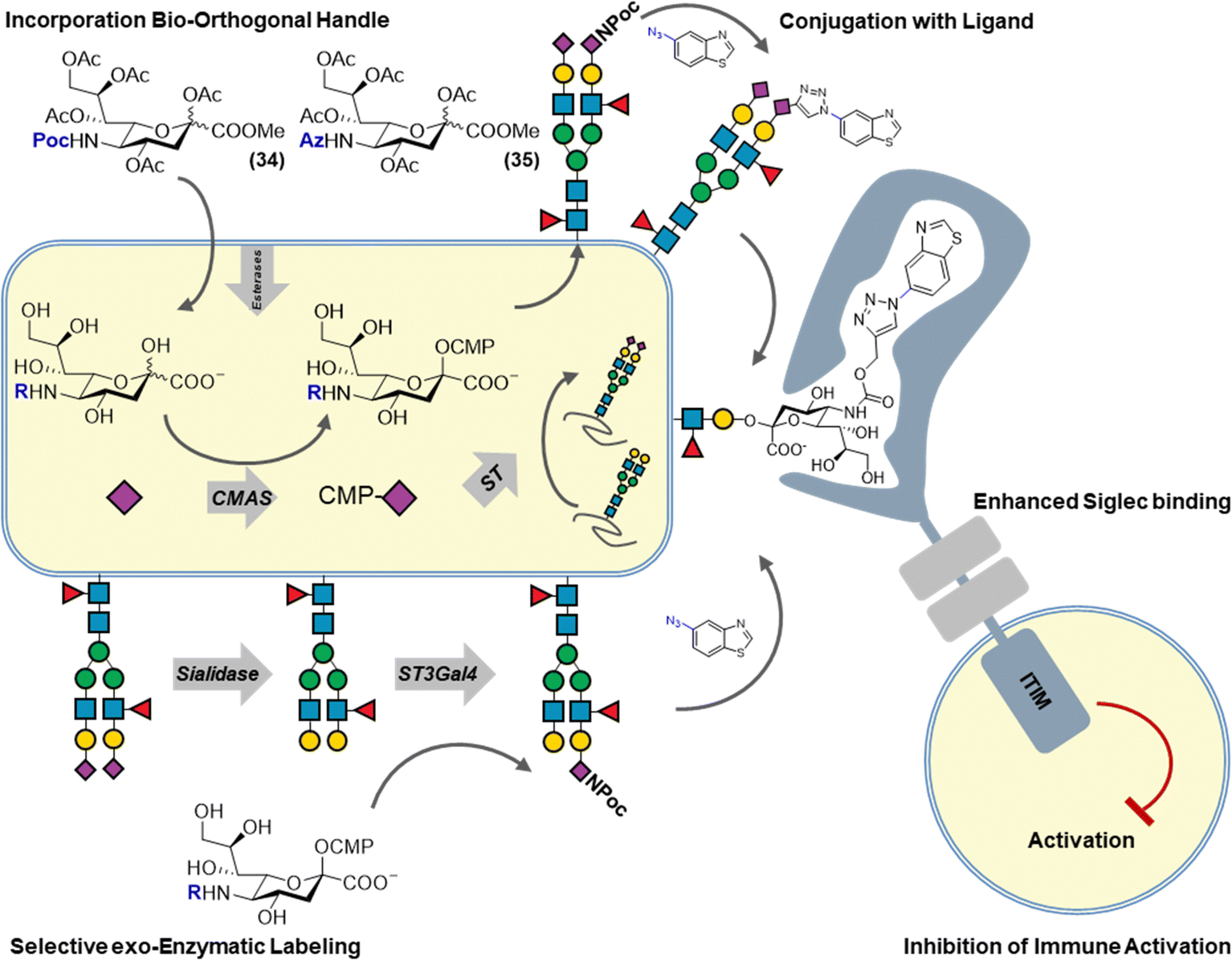 | ||
| Fig. 4 Metabolic labeling of cell surface sialic acid residues to introduce high affinity Siglec ligands using bioorthogonal chemistry. | ||
A potential drawback of our approach is the lack of control of the sialoglycan linkage type (α2,3 α2,6 α2,8) which contributes to the recognition of Siglec receptors. This hurdle has recently been overcome by the use of selective exoenzymatic labelling (SEEL) to introduce Siglec ligands (Fig. 4). Briard and co-workers used Chinese hamster ovary (CHO) cells with defined glycan isoforms expression that were chemoenzymatically engineered with alkyne or azide 6-modified fucoses and C-5 modified sialic acids. Several ligands were then added using bioorthogonal chemistry and Siglec binding was determined using flow cytometry. Cells expressing the newly found Siglec-15 ligands were able to impair the differentiation of osteoprogenitor cells to osteoclasts.47
Metabolic labelling reagents and inhibitors to study and alter host-pathogen interactions
Like mammalian cells, the bacterial cellular envelope is also covered in a dense layer of glycoconjugates. Many of these bacterial surface glycans, like lipopolysaccharide (LPS), capsular polysaccharide (CPS) and some glycosylated bacterial membrane proteins have been identified as key virulence factors and drive processes promoting bacterial infection such as immune evasion or suppression, resistance to antibodies and antimicrobial compounds, improved motility and increased biofilm formation. Bacteria also use glycans to adapt to their host and are known to express host-like glycans acting as a molecular cloak to escape immune surveillance. Antibodies raised against these bacterial host-like glycans are known to cross react with host glycans leading to autoimmune diseases like the Guillian–Barré syndrome. The human host immune system has adapted to express receptors that enable the detection of bacterial glycoconjugates for immune surveillance. Conversely, bacteria have evolved to express proteins to adhere and digest host glycans. Viruses reproduce their viral capsids in host cells and are known to be heavily glycosylated. Glycosylation of the viral capsid plays an important role viral infections as they interfere with immune-recognition. A prominent example is the glycosylation of the receptor binding domain (RBD) of the SARS-CoV-2 spike protein. Dense RDB glycosylation was shown to shield the protein from antibody recognition with the RBD only revealed upon conformational change when interacting with the ACE2 receptor.48–50 Another high profile role of viral glycosylation is the expression of high-mannose structures on the pg120 protein of the human immunodeficiency virus (HIV) which can serve as an antigen targeted by PGT121 type neutralizing antibodies.51,52To study glycans involved in the host pathogen interaction, it is important to understand the metabolic pathways leading to their biosynthesis. In humans, glycans are primarily composed of ten different monosaccharide building blocks which are biosynthesized through a complex metabolic network consisting of ∼40 enzymes and are the substrates for >170 glycosyltransferases enzymes. By comparison, bacteria express a much larger diversity of nucleotide sugars (∼70) and glycosyltranferases (>600) giving rise to an even more complex glycome. Furthermore, some bacteria express monosaccharide transporters to scavenge host derived monosaccharides (e.g. sialic acid) used as a carbon source or molecular mimicry purposes. By utilizing the differences in monosaccharide uptake and glycan biosynthesis between host and guest, the glycan mediated host pathogen interaction can be studied using metabolic labels and inhibitors.
Probes perturbing viral reproduction in host cells
In the case of modulating viral infections, this can be done by modifying the host-cell glycans. Reutter and co-workers established that cells treated the first unnatural mannosamine derivatives, ManNprop (18), ManNbut (19) and ManNpent (20), showed significantly changed susceptibility to infection when exposed to several viruses (Fig. 5).53 This was shown to be because of the effect of the unnatural modifications on the binding of the viral agglutinins to host cell sialylated glycans. Depending on the chain length and the type of virus, binding and subsequent infection could be decreased or even increased. For example, the incorporation of (18–20) led to a decrease in binding of murine and LPV primate polyoma viruses whilst in case of BKV primate polyoma binding was increased upon treatment of cells with the same analogs.54–56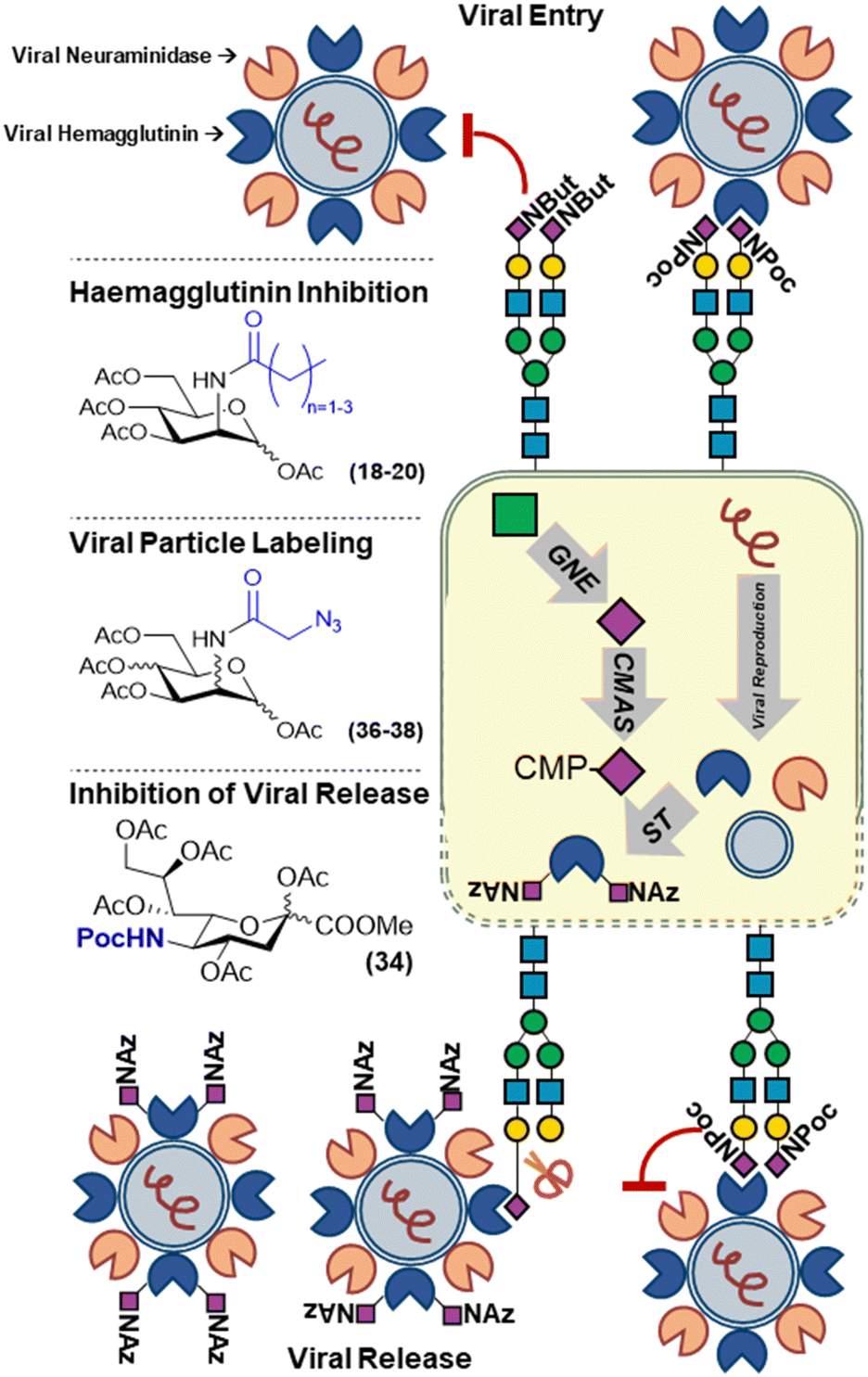 | ||
| Fig. 5 Metabolic labelling of sialic acids to track and perturb viral infections utilizing sialic acid expression. | ||
Recently, our group revisited this with sialic acid derivatives modified with bioorthogonal handles. The introduction of small chemical reporters on biomolecules is often expected to minimally impact their biological function and processing. However, we showed that incorporation of the unnatural sialic acid analogs can alter host-pathogen interactions. N-Propargyloxycarbonyl sialic acid (SiaNPoc 34), unlike N-azidoacetyl sialic acid (SiaNAz 35), both commonly used to metabolically label sialic acids on the cell membrane of mammalian cells, cannot be cleaved by several sialidases from different bacterial strains (Fig. 5). Moreover, cells grown with SiaNPoc (34) showed reduced viral reproduction upon infection with influenza virus. We hypothesized that the influenza virus hemagglutinin could still bind the sialic acids on host cells as initial infection rates between normal and SiaNPoc treated cells was similar. However, after viral reproduction inside the cells, the new viral particles could no longer be released by the viral neuraminidase, hence inhibiting progeny escape and the progression of infection.57 This underlines that unnatural modifications used to track biological processes involving modified sugars may not always be innocent bystanders, but can significantly alter these processes.
The utilization of the host glycosylation machinery by viruses was nicely demonstrated by Zhao et al. who showed that azidoacetyl hexosamine (ManNAz and GlcNAz) derivatives (36–37), were incorporated into viral capsid glycans (Fig. 5). Upon infection with the measles virus, newly formed viral particles could be labeled with azido sugars to track the cause of an infection.58 This strategy was later also used to label recombinant HIV gp120 glycoprotein using ManNAz (36) and GalNAz (38) and study the gp120 uptake by bone-marrow derived DC's.59
Probes to study and inhibit bacterial infections
Probes that selectively target bacterial glycosylation can be based on the unique sugars that they biosynthesize. For example, fluorinated nucleotide sugar derivatives such as CDP-6-deoxy-6-difluoroglucose (39) and ADP-2-fluoroheptose (40) have been shown to be good inhibitors of bacterial glycan biosynthesis enzymes but are not cell permeable (Fig. 6).29,60 Therefore, the synthesis of cell permeable metabolic precursors has been investigated to label or inhibit the expression of bacterial monosaccharides. An early example of this strategy is the use of 8-deoxy-8-azido-KDO (41), an analog of KDO, which is a bacteria-specific sugar important for LPS-biosynthesis. Incorporation of this azido analog led to both metabolic labelling of the LPS-core and truncation of the LPS in E. coli.61,62 Using peracetylated derivatives of bacterial monosaccharides bacillosamine (42), fucosamine (43) and 2,4-diaminofucose (dat, 44), glycosylation inhibitors targeting Helicobacter pylori glycans were developed by Dube, Kulkarni and co-workers.63 Similarly, Wennekes and co-workers reported the use of 2,4-diazidoacetyl-2,4-dideoxy-D-rhamnose (45) to label the expression of legionaminic acid sugars on Campylobacter Jenuni flagellin glycoproteins (Fig. 6). Using this metabolic label the function CMP–legionaminic acid transferase maf4 could be identified, by comparing incorporation between wild-type and Δmaf4-mutant C. jejuni.9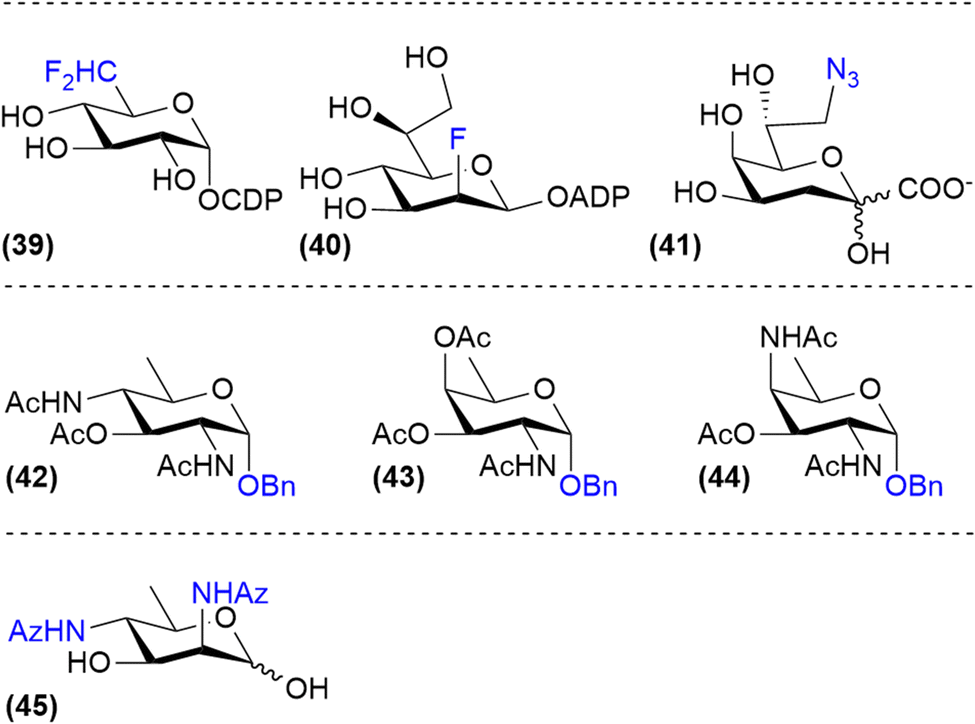 | ||
| Fig. 6 Metabolic labels and inhibitors based on bacterial monosaccharides. | ||
Hence, the use of peracetylated bacterial monosaccharide precursors enables the labelling of bacterial glycoconjugates akin to those reported for mammalian sugars. However, it should be noted that due to the much lower permeability of the bacterial envelope compared to mammalian cell membranes, millimolar concentrations of metabolic label are needed to achieve significant cell-membrane labelling in bacteria. This precludes the use of such strategies in more dilute conditions such as in vivo. To overcome this limitation, we investigated the use of bacterial monosaccharide transporters to achieve the efficient uptake of unnatural monosaccharides in bacteria.
Selective targeting of bacteria versus host cells
Due to the low permeability of the bacterial cellular envelope for acetylated sugars, metabolic probes to efficiently target these bacteria can be based on sugars that are actively transported over the membrane. Some bacteria have adapted to the human host to recognize and take-up host-derived monosaccharides such as sialic acid. A variety of transporters stemming from different transporter families have evolved to be able to transport sialic acids.64 In several pathogens, sialic acid uptake and utilization has been identified as a virulence factor.65 This has made sialic acid utilization an interesting target for the development of metabolic labels and inhibitors.For example, Neisseria gonorrhoeae cannot synthesize sialic acids itself, but can scavenge CMP–sialic acids from its surroundings and incorporate them on its LOS. N. gonorrhoeae causes a sexually transmitted disease, is increasingly resistant to antibiotics and sialic acid utilization has been identified as a virulence factor. Ram and co-workers investigated the use of modified CMP–sialic acid analogs (60–62) to glycoengineer N. gonorrhoeae and establish the effect on serum mediated killing (Fig. 7). The introduction of legionaminic acid by feeding CMP–legionaminic acid (60) led to a decrease in serum resistance and reduced the infection duration and burden in vivo.66 To a lesser extend reductions in serum resistance were achieved by incorporating KDN and 9-azidosialic acid by feeding their respective CMP-derivatives (61 and 62).67
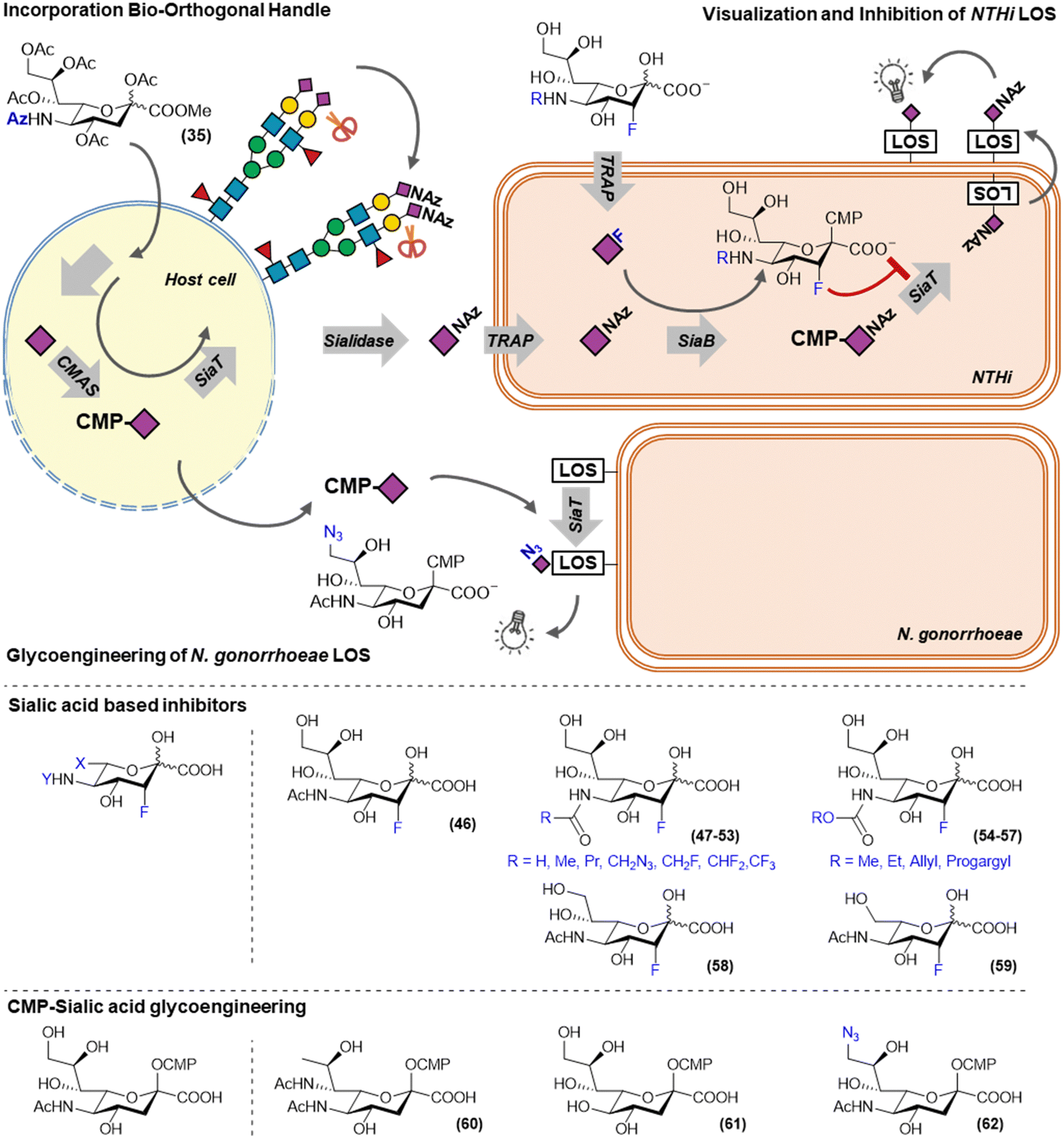 | ||
| Fig. 7 Metabolic labels and inhibitors to track and inhibit the sialic acid utilization in bacteria. | ||
Another bacterium that utilizes host derived sialic acids to support is growth and immune evasion is non-typeable Haemophilus influenzae (NTHi, Fig. 7). NTHi is a commensal Gram-negative bacterium that can act as an opportunistic pathogen in patients suffering from otitis media and chronic obstructive pulmonary disease (COPD). This switch to an opportunistic pathogen is associated with utilization of host sialic acids to decorate its lipooligosaccharide (LOS). LOS sialylation makes NTHi more resistant to serum mediated killing and increases biofilm formation.68,69 NTHi does not synthesize sialic acids itself and sequesters it from its environment using a TRAP-type transporter. As mammals are unable to actively take up sialic acid, our group recently showed that exploiting this difference in uptake, the NTHi sialic acid utilization pathway could be targeted selectively. We showed that unprotected sialic acid analogs were able to selectively label NTHi LOS, whereas peracetylated versions of these sialic acids selectively label the host-cell glycans.10 This utilization of host glycans was visualized in a co-culture model of PHBEC epithelial cells and NTHi. PHBEC were selectively fed peracetylated SiaNAz (35) via a trans-well setup and led to transfer of the azidosugars onto NTHi LOS. This host-to-NTHi sialic acid transfer was shown to be neuraminidase dependent as it could be inhibited by sialidase inhibitor DANA, or by incorporating sialidase resistant peracetylated SiaNPoc (34).57 It was also shown that feeding unprotected 3-fluorosialic acid selectively inhibited NTHi LOS sialylation at low concentrations, making it a potential therapeutic for people with otitis media or COPD that suffer from NTHi infections.10
In mammals, the potency of FNANA could be strongly improved by modifying the N-acetamide. Inspired by this, a small library of modified unprotected 3-fluorosialic acid derivatives was synthesized to explore the structure activity relationship for the NTHi sialic acid utilization enzymes.70 Unprotected unnatural sialic acid derivatives were chemo-enzymatically prepared from their respective mannosamine derivatives using recombinant N-acetyl neuraminate lyase (NAL).71,72 As this is an reversable reaction, an excess of one of the reagents was used to drive product formation. This is typically done in batch reactions, however, we also showed that NAL can be immobilized on beads without loss of enzymatic activity and remained active in a continuous flow setup for at least a week. With this strategy several N-modified sialic acids were synthesized (47–59).73 Besides modifications on the 5-position, also small modifications at the 9-positions were tested, as well as shortened glycerol tails. In vitro testing of these modified inhibitors on NTHi revealed that although small modifications at the C-5 and C-9 are tolerated, the enzymes processing the sialic acid in NTHi are much more restrictive than their mammalian counterparts.70
Recently, Ram and co-workers discovered that KDN (61) was also incorporated by NTHi. They also showed that humans produce anti-KDN antibodies, that are able to bind NTHi LOS decorated with KDN, and that the generation of those antibodies is similar to those generated against N-glycolylsialic acid. KDN was able to reduce NTHi infection in N-glycolylsialic acid deficient mice by decreasing the virulence related to host sialic acid incorporation. These two mechanisms make KDN a potential therapeutic option for NTHi infections.74
Conclusions
The field of metabolic glycoengineering has yielded many new probes to track and perturb glycosylation. The applications of these molecules are wide and range from novel cancer therapeutics to tools to modulate and study host-pathogen interactions.Author contributions
Text: E. R. and T. J. B.; figures: E. R., J. F. A. P. and T. J. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an ERC-Stg (GlycoEdit, 758913) awarded to TJB.Notes and references
- K. Ohtsubo and J. D. Marth, Cell, 2006, 126, 855–867 CrossRef PubMed.
- J. H. Prestegard, Essentials of glycobiology, Cold Spring Harbor Laboratory Press, New York, NY, United States, 3rd edn, 2017 Search PubMed.
- S. S. Pinho and C. A. Reis, Nat. Rev. Cancer, 2015, 15, 540–555 CrossRef PubMed.
- Y. Matsumoto, Trends Glycosci. Glycotechnol., 2021, 33, E33–E38 CrossRef.
- N. Rodrigues Mantuano, M. Natoli, A. Zippelius and H. Läubli, J. Immunother. Cancer, 2020, 8, e001222 CrossRef PubMed.
- H. Xiao, E. C. Woods, P. Vukojicic and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10304–10309 CrossRef CAS PubMed.
- T. J. Sminia, H. Zuilhof and T. Wennekes, Carbohydr. Res., 2016, 435, 121–141 CrossRef CAS PubMed.
- R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioconjugate Chem., 2021, 32, 2457–2479 CrossRef CAS PubMed.
- X. Meng, G.-J. Boons, M. M. S. M. Wösten and T. Wennekes, Angew. Chem., Int. Ed., 2021, 60, 24811–24816 CrossRef CAS PubMed.
- T. Heise, J. D. Langereis, E. Rossing, M. I. de Jonge, G. J. Adema, C. Büll and T. J. Boltje, Cell Chem. Biol., 2018, 25, 1279–1285.e1278 CrossRef CAS PubMed.
- D. H. Dube, K. Champasa and B. Wang, Chem. Commun., 2011, 47, 87–101 RSC.
- M. G. Alteen, H. Y. Tan and D. J. Vocadlo, Curr. Opin. Struct. Biol., 2021, 68, 157–165 CrossRef CAS PubMed.
- M. D. Burkart, S. P. Vincent, A. Düffels, B. W. Murray, S. V. Ley and C.-H. Wong, Bioorg. Med. Chem., 2000, 8, 1937–1946 CrossRef CAS PubMed.
- C. D. Rillahan, A. Antonopoulos, C. T. Lefort, R. Sonon, P. Azadi, K. Ley, A. Dell, S. M. Haslam and J. C. Paulson, Nat. Chem. Biol., 2012, 8, 661–668 CrossRef CAS PubMed.
- Y. Zhou, T. Fukuda, Q. Hang, S. Hou, T. Isaji, A. Kameyama and J. Gu, Sci. Rep., 2017, 7, 11563 CrossRef PubMed.
- M. A. Carrascal, M. Silva, J. S. Ramalho, C. Pen, M. Martins, C. Pascoal, C. Amaral, I. Serrano, M. J. Oliveira, R. Sackstein and P. A. Videira, Mol. Oncol., 2018, 12, 579–593 CrossRef CAS PubMed.
- N. M. Okeley, S. C. Alley, M. E. Anderson, T. E. Boursalian, P. J. Burke, K. M. Emmerton, S. C. Jeffrey, K. Klussman, C.-L. Law, D. Sussman, B. E. Toki, L. Westendorf, W. Zeng, X. Zhang, D. R. Benjamin and P. D. Senter, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5404–5409 CrossRef CAS PubMed.
- N. M. Okeley, R. A. Heiser, W. Zeng, S. M. Hengel, J. Wall, P. C. Haughney, T. A. Yap, F. Robert, R. E. Sanborn, H. Burris, L. Q. Chow, K. T. Do, M. Gutierrez, K. Reckamp, A. Weise, D. R. Camidge, J. Strickler, C. Steuer, Z. Wang, M. M. O'Meara, S. C. Alley and S. J. Gardai, Cancer Res., 2018, 78, 5551 CrossRef.
- M. L. Disis, L. R. Corulli, E. A. Gad, M. R. Koehnlein, D. L. Cecil, P. D. Senter, S. J. Gardai and N. M. Okeley, Mol. Cancer Ther., 2020, 19(5), 1102–1109 CrossRef CAS PubMed.
- K. T. Do, L. Q. M. Chow, K. Reckamp, R. E. Sanborn, H. Burris, F. Robert, D. R. Camidge, C. E. Steuer, J. H. Strickler, A. Weise, J. M. Specht, M. Gutierrez, P. Haughney, S. Hengel, C. L. Derleth and T. A. Yap, Oncologist, 2021, 26, 925-e1918 CrossRef PubMed.
- J. G. Allen, M. Mujacic, M. J. Frohn, A. J. Pickrell, P. Kodama, D. Bagal, T. San Miguel, E. A. Sickmier, S. Osgood, A. Swietlow, V. Li, J. B. Jordan, K.-W. Kim, A.-M. C. Rousseau, Y.-J. Kim, S. Caille, M. Achmatowicz, O. Thiel, C. H. Fotsch, P. Reddy and J. D. McCarter, ACS Chem. Biol., 2016, 11, 2734–2743 CrossRef CAS PubMed.
- Y. Dai, R. Hartke, C. Li, Q. Yang, J. O. Liu and L.-X. Wang, ACS Chem. Biol., 2020, 15, 2662–2672 CrossRef CAS PubMed.
- T. M. Gloster, W. F. Zandberg, J. E. Heinonen, D. L. Shen, L. Deng and D. J. Vocadlo, Nat. Chem. Biol., 2011, 7, 174–181 CrossRef CAS PubMed.
- M. Zimmermann, M. Nguyen, C. M. Schultheiss, H. Kolmar and A. Zimmer, Biotechnol. Bioeng., 2021, 118, 1818–1831 CrossRef CAS PubMed.
- Y. Kizuka, M. Nakano, Y. Yamaguchi, K. Nakajima, R. Oka, K. Sato, C. T. Ren, T. L. Hsu, C. H. Wong and N. Taniguchi, Cell Chem. Biol., 2017, 24, 1467–1478 e1465 CrossRef CAS PubMed.
- C. Ma, H. Takeuchi, H. Hao, C. Yonekawa, K. Nakajima, M. Nagae, T. Okajima, R. S. Haltiwanger and Y. Kizuka, Int. J. Mol. Sci., 2020, 21, 6007 CrossRef CAS PubMed.
- J. F. A. Pijnenborg, E. A. Visser, M. Noga, E. Rossing, R. Veizaj, D. J. Lefeber, C. Büll and T. J. Boltje, Chem. – Eur. J., 2021, 27, 4022–4027 CrossRef CAS PubMed.
- P. D. Yurchenco and P. H. Atkinson, Biochemistry, 1975, 14, 3107–3114 CrossRef CAS PubMed.
- C.-W. T. Chang, X. H. Chen and H.-W. Liu, J. Am. Chem. Soc., 1998, 120, 9698–9699 CrossRef CAS.
- J. F. A. Pijnenborg, E. Rossing, J. Merx, M. J. Noga, W. H. C. Titulaer, N. Eerden, R. Veizaj, P. B. White, D. J. Lefeber and T. J. Boltje, Nat. Commun., 2021, 12, 7024 CrossRef PubMed.
- L. K. Mahal, N. W. Charter, K. Angata, M. Fukuda, D. E. Koshland and C. R. Bertozzi, Science, 2001, 294, 380–381 CrossRef PubMed.
- R. Horstkorte, M. Mühlenhoff, W. Reutter, S. Nöhring, M. Zimmermann-Kordmann and R. Gerardy-Schahn, Exp. Cell Res., 2004, 298, 268–274 CrossRef PubMed.
- M. Nagasundaram, R. Horstkorte and V. S. Gnanapragassam, Molecules, 2020, 25(11), 2632 CrossRef PubMed.
- P. R. Wratil, S. Rigol, B. Solecka, G. Kohla, C. Kannicht, W. Reutter, A. Giannis and L. D. Nguyen, J. Biol. Chem., 2014, 289, 32056–32063 CrossRef PubMed.
- O. Nieto-Garcia, P. R. Wratil, L. D. Nguyen, V. Böhrsch, S. Hinderlich, W. Reutter and C. P. R. Hackenberger, Chem. Sci., 2016, 7, 3928–3933 RSC.
- C. Büll, T. J. Boltje, M. Wassink, A. M. A. de Graaf, F. L. van Delft, M. H. den Brok and G. J. Adema, Mol. Cancer Ther., 2013, 12, 1935–1946 CrossRef PubMed.
- N. Alessandro, L. F. Mariah, H. Sophie, F. Carolyne, K.-M. Lucy, S. M. Matthew, R. R. Michaela and O. D. Michael, Haematologica, 2020, 105, 457–467 CrossRef PubMed.
- J. Daly, S. Sarkar, A. Natoni, J. C. Stark, N. M. Riley, C. R. Bertozzi, M. Carlsten and M. E. O’Dwyer, Blood Adv., 2022, 6(11), 3352–3366 CrossRef CAS PubMed.
- C. Büll, T. J. Boltje, E. A. W. Van Dinther, T. Peters, A. M. A. De Graaf, J. H. W. Leusen, M. Kreutz, C. G. Figdor, M. H. den Brok and G. J. Adema, ACS Nano, 2015, 9, 733–745 CrossRef PubMed.
- C. Büll, E. Collado-Camps, E. D. Kers-Rebel, T. Heise, J. N. Søndergaard, M. H. den Brok, B. M. Schulte, T. J. Boltje and G. J. Adema, Immunol. Cell Biol., 2017, 95, 408–415 CrossRef PubMed.
- L. J. Edgar, A. J. Thompson, V. F. Vartabedian, C. Kikuchi, J. L. Woehl, J. R. Teijaro and J. C. Paulson, ACS Cent. Sci., 2021, 7, 1508–1515 CrossRef CAS PubMed.
- N. Balneger, L. A. M. Cornelissen, M. Wassink, S. J. Moons, T. J. Boltje, Y. E. Bar-Ephraim, K. K. Das, J. N. Søndergaard, C. Büll and G. J. Adema, Cell. Mol. Life Sci., 2022, 79, 98 CrossRef CAS PubMed.
- S. J. Moons, G. J. Adema, M. T. G. M. Derks, T. J. Boltje and C. Büll, Glycobiology, 2019, 29, 433–445 CAS.
- T. Heise, J. F. A. Pijnenborg, C. Büll, N. van Hilten, E. D. Kers-Rebel, N. Balneger, H. Elferink, G. J. Adema and T. J. Boltje, J. Med. Chem., 2019, 62, 1014–1021 CrossRef CAS PubMed.
- S. J. Moons, E. Rossing, M. A. C. H. Janssen, T. Heise, C. Büll, G. J. Adema and T. J. Boltje, ACS Chem. Biol., 2022, 17, 590–597 CrossRef CAS PubMed.
- C. Büll, T. Heise, N. van Hilten, J. F. A. Pijnenborg, V. R. L. J. Bloemendal, L. Gerrits, E. D. Kers-Rebel, T. Ritschel, M. H. den Brok, G. J. Adema and T. J. Boltje, Angew. Chem., Int. Ed., 2017, 56, 3309–3313 CrossRef PubMed.
- J. G. Briard, H. Jiang, K. W. Moremen, M. S. Macauley and P. Wu, Nat. Commun., 2018, 9, 880 CrossRef PubMed.
- L. Casalino, Z. Gaieb, J. A. Goldsmith, C. K. Hjorth, A. C. Dommer, A. M. Harbison, C. A. Fogarty, E. P. Barros, B. C. Taylor, J. S. McLellan, E. Fadda and R. E. Amaro, ACS Cent. Sci., 2020, 6, 1722–1734 CrossRef CAS PubMed.
- T. Sztain, S.-H. Ahn, A. T. Bogetti, L. Casalino, J. A. Goldsmith, E. Seitz, R. S. McCool, F. L. Kearns, F. Acosta-Reyes, S. Maji, G. Mashayekhi, J. A. McCammon, A. Ourmazd, J. Frank, J. S. McLellan, L. T. Chong and R. E. Amaro, Nat. Chem., 2021, 13, 963–968 CrossRef CAS PubMed.
- O. C. Grant, D. Montgomery, K. Ito and R. J. Woods, Sci. Rep., 2020, 10, 14991 CrossRef PubMed.
- M. Raska, K. Takahashi, L. Czernekova, K. Zachova, S. Hall, Z. Moldoveanu, M. C. Elliott, L. Wilson, R. Brown, D. Jancova, S. Barnes, J. Vrbkova, M. Tomana, P. D. Smith, J. Mestecky, M. B. Renfrow and J. Novak, J. Biol. Chem., 2010, 285, 20860–20869 CrossRef CAS PubMed.
- J.-P. Julien, D. Sok, R. Khayat, J. H. Lee, K. J. Doores, L. M. Walker, A. Ramos, D. C. Diwanji, R. Pejchal, A. Cupo, U. Katpally, R. S. Depetris, R. L. Stanfield, R. McBride, A. J. Marozsan, J. C. Paulson, R. W. Sanders, J. P. Moore, D. R. Burton, P. Poignard, A. B. Ward and I. A. Wilson, PLoS Pathog., 2013, 9, e1003342 CrossRef CAS PubMed.
- O. T. Keppler, R. Horstkorte, M. Pawlita, C. Schmidt and W. Reutter, Glycobiology, 2001, 11, 11R–18R CrossRef CAS PubMed.
- O. T. Keppler, P. Stehling, M. Herrmann, H. Kayser, D. Grunow, W. Reutter and M. Pawlita, J. Biol. Chem., 1995, 270, 1308–1314 CrossRef CAS PubMed.
- M. Herrmann, C. W. von der Lieth, P. Stehling, W. Reutter and M. Pawlita, J. Virol., 1997, 71, 5922–5931 CrossRef CAS PubMed.
- O. T. Keppler, M. Herrmann, C. W. von der Lieth, P. Stehling, W. Reutter and M. Pawlita, Biochem. Biophys. Res. Commun., 1998, 253, 437–442 CrossRef CAS PubMed.
- T. Heise, C. Büll, D. M. Beurskens, E. Rossing, M. I. de Jonge, G. J. Adema, T. J. Boltje and J. D. Langereis, Bioconjugate Chem., 2017, 28, 1811–1815 CrossRef CAS PubMed.
- X. Zhao, L. Cai, E. A. Adogla, H. Guan, Y. Lin and Q. Wang, Bioconjugate Chem., 2015, 26, 1868–1872 CrossRef CAS PubMed.
- L. Sun, M. Ishihara, D. R. Middleton, M. Tiemeyer and F. Y. Avci, J. Biol. Chem., 2018, 293, 15178–15194 CrossRef CAS PubMed.
- S. Grizot, M. Salem, V. Vongsouthi, L. Durand, F. Moreau, H. Dohi, S. Vincent, S. Escaich and A. Ducruix, J. Mol. Biol., 2006, 363, 383–394 CrossRef CAS PubMed.
- A. Dumont, A. Malleron, M. Awwad, S. Dukan and B. Vauzeilles, Angew. Chem., Int. Ed., 2012, 51, 3143–3146 CrossRef PubMed.
- I. Nilsson, K. Grove, D. Dovala, T. Uehara, G. Lapointe and D. A. Six, J. Biol. Chem., 2017, 292, 19840–19848 CrossRef PubMed.
- D. A. Williams, K. Pradhan, A. Paul, I. R. Olin, O. T. Tuck, K. D. Moulton, S. S. Kulkarni and D. H. Dube, Chem. Sci., 2020, 11, 1761–1774 RSC.
- Gavin H. Thomas, Biochem. Soc. Trans., 2016, 44, 760–765 CrossRef PubMed.
- E. Severi, D. W. Hood and G. H. Thomas, Microbiology, 2007, 153, 2817–2822 CrossRef PubMed.
- S. Gulati, I. C. Schoenhofen, D. M. Whitfield, A. D. Cox, J. Li, F. Michael, E. V. Vinogradov, J. Stupak, B. Zheng, M. Ohnishi, M. Unemo, L. A. Lewis, R. E. Taylor, C. S. Landig, S. Diaz, G. W. Reed, A. Varki, P. A. Rice and S. Ram, PLoS Pathog., 2015, 11, e1005290 CrossRef PubMed.
- S. Gulati, I. C. Schoenhofen, T. Lindhout-Djukic, M. J. Schur, C. S. Landig, S. Saha, L. Deng, L. A. Lewis, B. Zheng, A. Varki and S. Ram, J. Immunol., 2020, 204, 3283 CrossRef PubMed.
- T. Hallström and K. Riesbeck, Trends Microbiol., 2010, 18, 258–265 CrossRef PubMed.
- J. D. Langereis and P. W. M. Hermans, FEMS Microbiol. Lett., 2013, 346, 81–89 CrossRef CAS PubMed.
- S. J. Moons, E. Rossing, J. J. A. Heming, M. A. C. H. Janssen, M. van Scherpenzeel, D. J. Lefeber, M. I. de Jonge, J. D. Langereis and T. J. Boltje, Bioconjugate Chem., 2021, 32, 1047–1051 CrossRef CAS PubMed.
- P. Laborda, S.-Y. Wang, A.-M. Lu, M. He, X.-C. Duan, Y.-J. Qian, Y.-S. Jung, L. Liu and J. Voglmeir, Adv. Synth. Catal., 2017, 359, 3120–3125 CrossRef CAS.
- Y. Tanaka and J. J. Kohler, J. Am. Chem. Soc., 2008, 130, 3278–3279 CrossRef CAS PubMed.
- V. R. L. J. Bloemendal, S. J. Moons, J. J. A. Heming, M. Chayoua, O. Niesink, J. C. M. Van Hest, T. J. Boltje and F. P. J. T. Rutjes, Adv. Synth. Catal., 2019, 361, 2443–2447 CrossRef CAS PubMed.
- S. Saha, A. Coady, A. Sasmal, K. Kawanishi, B. Choudhury, H. Yu, U. Sorensen Ricardo, J. Inostroza, C. Schoenhofen Ian, X. Chen, A. Münster-Kühnel, C. Sato, K. Kitajima, S. Ram, V. Nizet, A. Varki, F. Feldman Mario and L. Doering Tamara, mBio, 2021, 12, e03226-03220 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |