
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA neutral vicinal silylene/phosphane supported six-membered C2PSiAu2 ring and a silver(I) complex†
Mohd
Nazish
 a,
Han
Bai
b,
Christina M.
Legendre
a,
Regine
Herbst-Irmer
a,
Lili
Zhao
*b,
Dietmar
Stalke
*a and
Herbert W.
Roesky
*a
a,
Han
Bai
b,
Christina M.
Legendre
a,
Regine
Herbst-Irmer
a,
Lili
Zhao
*b,
Dietmar
Stalke
*a and
Herbert W.
Roesky
*a
aInstitut für Anorganische Chemie, Universität Göttingen, Tammannstrasse 4, Göttingen, 37077, Germany. E-mail: hroesky@gwdg.de
bInstitute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing, 211816, China
First published on 24th October 2022
Abstract
This work presents the different coordination nature of the bidentate ligand towards gold and silver complexes. The reaction of 1 with AuClSMe2 in dichloromethane resulted in two gold atoms containing six-membered ring PhC(NtBu)2Si–Au⋯Au–PPh2C6H4 (2). Compound 2 exhibits intramolecular aurophilic interaction (2.9987(7) Å), which is further supported by quantum chemical calculations. Moreover, the reduction of aluminium adduct 3 with AgSbF6 affords insertion of silver cation [{PhC(NtBu)2SiF2}-C6H4(PPh2)-Ag-(PPh2)C6H4{PhC(NtBu)2SiF2}]AlCl4− (4) between two phosphane. In compound 4 only two P(III) of two molecules of 1 coordinates to Ag(I), while two Si(II) remains uncoordinated and gives oxidative addition of Fluorine.
Recent facile and high yield isolation of functionalized Si(II) compounds such as [PhC(NtBu)2SiCl]1 and [PhC(NtBu)2SiR]2 has led to an emerging area of chemistry centered around their coordination properties towards transition metal complexes.3,4 The propensity of silylenes to donate their lone pair to a decorated transition metal moiety led to the isolation of a range of silylene–transition metal complexes.4 Nevertheless, the silylene–gold complex is highly unprecedented and there are only scarce reports of silylene–Au(I) complexes that feature an Au–Au (aurophilic) interaction.5a–e Aurophilic interactions range roughly from a distance of 2.5 to 3.5 Å and It is exploited as a tool for the synthesis of several unusual multinuclear gold complexes, which exhibit intriguing electronic absorption and luminescence properties.5f–t A benzamidinato stabilized silylene gold complex [{PhC(NtBu)2}Si{N(SiMe3)2}]AuCl was already isolated.6 Obviously the ligand does not exhibit any bidentate nature along with Au–Au interaction. Furthermore, the hydrosilylation of ketone, reduction of amide, catalytic borylation of arene, and C–C bond formation reactions were described as mediated by various NHSi–transition-metal complexes.7 To the best of our knowledge, NHSi–coinage-metal complexes are still very scant, despite the theoretical impetus confirming that NHSi forms strong bonds with the hosted metals.8 The trend of the silylene–coinage-metal bond strength is as follows: Au > Cu > Ag. Prior to theoretical investigations, a Fischer-type Si(II)–Au complex [(η5-Me5C5)(η1-Me5C5)Si
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) AuCl] was isolated from the reaction of [(η5-Me5C5)2Si] with Au(CO)Cl under elimination of CO.9 The insertion of [(η5-Me5C5)2Si] into a Au–Cl bond, resulting in a silyl gold complex, was also reported in 2016.10a Starting from this state of knowledge, the bidentate neutral ligand PhC(NtBu)2Si-C6H4(PPh2) (1)11 should be advantageous to bring coinage metals in close proximity. The main feature of 1 is the simultaneous presence of the donor sites Si: and P:. Given the paucity of NHSi–coinage-metal complexes, we explored the reaction of 1 with the easily available Group 11 metal salts, e.g. AuCl(SMe2) and AgSbF6. In order to expand the study of the silylene–phosphane ligand with Ag and Au complex, we first reacted a Si(II) ligand (1) with AuCl(SMe2) to form a complex PhC(NtBu)2Si–Au⋯Au–PPh2C6H4 (2). Dehalogenation of 3 [{PhC(NtBu)2}Si-C6H4(PPh2)AlCl3] with AgSbF6 gives an unexpected complex with the silver atom coordinated exclusively by the phosphorus sites of two ligands, for their part difluorinated at the silylene site. This leaves the ligand still neutral and rises the silicon oxidation state to (IV). The AlCl3 moiety from the starting material forms the single AlCl4− anion to the decorated silver cation in [{PhC(NtBu)2SiF2}-C6H4(PPh2)-Ag-(PPh2)C6H4{PhC(NtBu)2SiF2}]AlCl4− (4).
AuCl] was isolated from the reaction of [(η5-Me5C5)2Si] with Au(CO)Cl under elimination of CO.9 The insertion of [(η5-Me5C5)2Si] into a Au–Cl bond, resulting in a silyl gold complex, was also reported in 2016.10a Starting from this state of knowledge, the bidentate neutral ligand PhC(NtBu)2Si-C6H4(PPh2) (1)11 should be advantageous to bring coinage metals in close proximity. The main feature of 1 is the simultaneous presence of the donor sites Si: and P:. Given the paucity of NHSi–coinage-metal complexes, we explored the reaction of 1 with the easily available Group 11 metal salts, e.g. AuCl(SMe2) and AgSbF6. In order to expand the study of the silylene–phosphane ligand with Ag and Au complex, we first reacted a Si(II) ligand (1) with AuCl(SMe2) to form a complex PhC(NtBu)2Si–Au⋯Au–PPh2C6H4 (2). Dehalogenation of 3 [{PhC(NtBu)2}Si-C6H4(PPh2)AlCl3] with AgSbF6 gives an unexpected complex with the silver atom coordinated exclusively by the phosphorus sites of two ligands, for their part difluorinated at the silylene site. This leaves the ligand still neutral and rises the silicon oxidation state to (IV). The AlCl3 moiety from the starting material forms the single AlCl4− anion to the decorated silver cation in [{PhC(NtBu)2SiF2}-C6H4(PPh2)-Ag-(PPh2)C6H4{PhC(NtBu)2SiF2}]AlCl4− (4).
Treatment of ligand 1 with AuCl(SMe2) in dichloromethane at room temperature for 20 hours resulted in 2 as a six-membered C–Si–Au⋯Au–P–C ring. One of the introduced AuCl units coordinates to the silylene and the other to the phosphane site of the ligand to give an Au–Au bond (Scheme 1).10a Compound 2 was obtained as colorless crystals at −30 °C from a concentrated solution of dichloromethane in 76% yield after two months (Scheme 1). The phosphane–silylene coordination to the Au atoms was verified by various NMR spectroscopic experiments (1H, 13C, 31P, 29Si) (Fig. S1–S4, ESI†). Compound 2 displays one resonance in the 1H NMR spectrum for two tertiary butyl groups at the silylene site at 1.04 ppm which is slightly upfield shifted when compared to ligand 1 (1.11 ppm). A single resonance at 25.57 (s) ppm in the 31P NMR spectrum, shifted downfield in comparison to that of 1 (−11.18 ppm), indicating the P→Au coordination. The 29Si NMR spectrum of 2 displays one doublet resonance at 1.97 ppm (JSi–P = 45.5 Hz), which corresponds to the Si(II) atom showing an upfield shift in comparison to 1 (18.52 ppm). Additionally, the formation of 2 was confirmed by LIFDI mass spectrometry in a toluene solution. It exhibits the molecular ion signal at m/z 984.09, corresponding to the formation of the gold adduct 2 (Fig. S5, ESI†). It melts at 245–250 °C as determined by differential scanning calorimetry.
 | ||
| Scheme 1 Synthesis of compound 2. | ||
Single crystals of 2 were characterized by X-ray diffraction (Fig. 1). The complex crystallizes in the monoclinic space group P21/n with one molecule and one not fully occupied dichloromethane molecule per asymmetric unit. The data were collected on a split crystal treated as a three-component twin. The X-ray diffraction analysis revealed that ligand 1 coordinates two gold atoms, one on its silylene site, and the other on the phosphane site. Each gold atom Au(I) is further coordinated by a chlorine atom in an almost ideally linear arrangement (173.86(13) and 175.42(12)°). The intramolecular Au–Au bond was found to be 2.9987(7) Å long, which is in the typical range for aurophilic bonds, resulting in a six-membered C–Si–Au–Au–P–C ring. The tendency for Au atoms to aggregate might have helped the formation of complex 2, which was obtained in high yields. No intermolecular aurophilic bonds were observed, as the distance between two gold atoms of two adjacent molecules is rather large (>7.8 Å). Quantum chemical calculations were performed to gain insight into the bonding nature of complex 2. As detailed in Fig. S14 (ESI†), the optimized structure of 2 agrees well with the experimental crystal structure, and it has the singlet electronic ground state, which is 51.3 kcal mol−1 lower in energy than the corresponding triplet state. The Au⋯Au aurophilic bond character can be further demonstrated by the QTAIM (Quantum Theory of Atoms in Molecules) analysis, as shown by the bond path (black line) and a bond critical point (green dot) between the gold atoms in Fig. 2. The electron density at the Au⋯Au bond critical point is only 0.03 e Å−3 and hence much lower than e.g. at the Mn–Mn bond critical point of (OC)5Mn–Mn(CO)5 at 0.19 e Å−310b or at the non-polar mid-point between the two manganese atoms in the dimetallaborane [tBuB{Mn(CO)3}2] of 0.22 e Å−3.10c
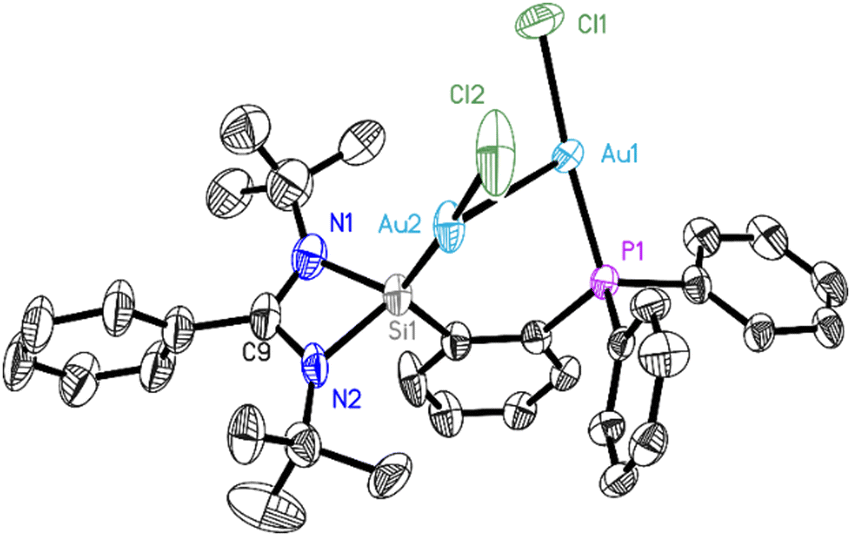 | ||
| Fig. 1 Molecular structure of 2. The anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms, solvent molecules, and minor components of disordered groups are omitted for clarity. Selected experimental [and at the BP86 + D3(BJ)/def2-TZVP(SMD, solvent = toluene) level calculated] bond lengths [Å] and angles [°]: Au1–P1 2.228(3) [2.249], Au1–Cl1 2.287(3) [2.318], Au1–Au2 2.9987(7) [3.039], Au2–Si1 2.238(3) [2.255], Au2–Cl2 2.325(4) [2.353], Si1–N2 1.812(9) [1.843], Si1–N1 1.815(9) [1.839],P1–Au1–Cl1 175.42(12) [174.7], P1–Au1–Au2 85.25(7) [83.6], Cl1–Au1–Au2 98.34(10) [101.6], Si1–Au2–Cl2 173.86(13) [173.6], Si1–Au2–Au1 87.18(8) [81.8], Cl2–Au2–Au1 98.85(13) [104.2]. | ||
 | ||
| Fig. 2 Isosurface map of interaction region indicator of 2 (top). Laplacian distribution of 2 according to plane P–Au–Au at the BP86/def2-TZVP(SMD, solvent = toluene) level (bottom). Red lines indicate areas of charge concentration (∇2ρ(r) < 0) while blue lines show areas of charge depletion (∇2ρ(r) > 0). The solid lines connecting the atomic nuclei are the bond paths. Green dots are bond critical points (BCPs). | ||
More detailed information about the Si–Au and P–Au bonds in molecule 2 is available from state-of-the-art energy decomposition analysis with natural orbitals for chemical valence (EDA-NOCV) method (see Computational Detail Section). We considered the interacting fragments in various electronic states and found the most appropriate fragments are best described as the neutral 1 and 2AuCl species in their electronic singlet states (Table S4, ESI†) because of their smaller stabilizing orbital interactions energy ΔEorb. The contribution of the orbital interaction term, ΔEorb (27.9%), is much weaker than the electrostatic attraction energy, ΔEelstat (66.4%). The dispersion forces provide the remaining 5.7% of the total attraction. The most important information about orbital interactions stems from the breakdown of the ΔEorb term into pairwise orbital contributions. Three major orbital interactions can be identified by inspecting the deformation densities Δρ1–Δρ3 associated with ΔEorb1–ΔEorb3 in Fig. S15 (ESI†). The strongest orbital interaction, ΔEorb1 (−58.0 kcal mol−1), comes from the σ-donation of the silylene Si-atom to one Au-atom of AuCl, giving the Si→Au dative bond. The second-largest interaction, ΔEorb2 (−44.2 kcal mol−1), is mainly due to the σ-donation from the phosphane P-atom to the Au-atom of the other AuCl moiety, generating the P→Au dative bond. Therefore, the silylene and phosphane moieties play synergistic roles in the coordination chemistry to Au in complex 2. The third contribution Δρ3 arises mainly from the π-backdonation of the AuCl part to the silylene and phosphane sites, as well as Au⋯Au orbital-orbital interactions, which cannot be strictly separated. The detailed individual components and most important interacting MOs of the neutral fragments (Fig. S16, ESI†). For comparison, we also analyzed the bonding nature of the bis(phosphane) analogs. The most appropriate interacting fragments for the bis(phosphane) analogs 2P–2AuCl are best described as the neutral 2P and 2AuCl species (Table S6, ESI†), which is similar to that of complex 2. There are two major orbital interactions identifying as the deformation densities Δρ1–Δρ2 associated with ΔEorb1–ΔEorb2 (Fig. S17, ESI†). The strongest orbital interaction, ΔEorb1 (−89.1 kcal mol−1), comes mainly from the σ-donation of P-atoms to the Au-atoms, making the two P→Au dative bonds. The second contribution Δρ2 is resembling the Δρ3 of complex 2 (Fig. S18, ESI†).
Phosphanes containing ligands are particularly appropriate for stabilizing low-valent metal ions. Consequently, there are numerous diphosphane containing silver(I) complexes.12 Recently these complexes have attracted considerable attention because of their applications in several homogeneous catalytic processes.13a Some of these complexes have also shown antitumor activity, as well as antifungal and antibacterial properties.13b,c In 2011 Bourissou et al. presented a synthesis of an Ag(I) cation stabilized by diphosphane.14 Similarly, using ligand 1, which features both a phosphane and a silylene donating site. We synthesized a silver cation stabilized by two phosphane groups, along with oxidative fluorination of the silylene by simultaneous reduction of Sb(V) to Sb(0) as a black precipitate. We used the silylene phosphane stabilized Al(III) adduct (3)11 as a starting material for the reaction with silver hexafluoro antimonate (AgSbF6) in dichloromethane at room temperature. After overnight stirring, the solution was filtered and concentrated under reduced pressure. After 1 month, colorless crystals of a silver complex [L1AgL1]+[AlCl4]− (L1= PPh2C6H4SiF2L) (4) were obtained at 0 °C from a concentrated solution of dichloromethane in 60% yield (Scheme 2).
 | ||
| Scheme 2 Synthetic route for the synthesis of compound 4. | ||
Crystals of 4 were subsequently analyzed by single-crystal X-ray diffraction, presented in Fig. 3.15 Compound 4 crystallizes in the monoclinic space group P21/c. Interestingly, instead of using the silylene pockets, the two ligands of 1 are stabilizing the silver atom through the phosphane donor sites. The Ag(I) cation is in a bent environment, featuring a P–Ag–P angle of 157.06(3)° and Ag–P distances of 2.4350(10) (P1) and 2.4291(8) Å (P3), respectively. The silylene moieties are oxidized by two fluorine atoms each, featuring a long Si–F (1.66 Å) and a short Si–F bond (1.61 Å). This results in elongated Si–N bonds (Si1–N1 1.801(3), Si1–N2 1.976(3), Si2–N4 1.823(3), Si2–N3 1.951(3) Å in 4, compared to Si1–N2 1.812(9), Si1–N1 1.815(9) Å in 2) giving a distorted square pyramidal environment around the penta-coordinated silicon atoms.
 | ||
| Fig. 3 Crystal structure of 4. Thermal ellipsoids are represented at the 50% probability level. Hydrogen atoms, solvent molecules, and minor components of disordered groups are omitted. Selected bond lengths [Å] and angles [°]: Ag1–P3 2.4291(8), Ag1–P1 2.4350(10), Si1–F1 1.606(2), Si1–F2 1.6577(19), Si1–N1 1.801(3), Si1–N2 1.976(3), Si2–F3 1.606(2), Si2–F4 1.654(2), Si2–N4 1.823(3), Si2–N3 1.951(3), P3–Ag1–P1 157.06(3). | ||
Compound 4 is further characterized by 1H, 13C, 31P, and 19F NMR spectroscopy (Fig. S6–S10, ESI†). The 29Si NMR spectrum in CD2Cl2 exhibits one triplet resonance at δ −97.76 ppm (JSi–F = 255 Hz) for the Si(IV) atoms, which is in good agreement with pentacoordinate silicon complexes.16 The 1H NMR spectrum of 4 displays resonances for tBu (δ 0.79 ppm) and for phenyl protons (δ 7.21–7.94 ppm) which are fully consistent with their solid-state molecular structure established by single-crystal X-ray diffraction. The 31P NMR spectrum shows two resonances at δ 9.12 and 11.93 ppm (sextet, JP–C = 25 Hz), which are shifted downfield in comparison to the neat ligand.11 The appearance of one quartet signal at δ −107.98 ppm (JSi–F = 150 Hz) for the fluorine atoms in the 19F NMR spectrum indicates the two fluorine atoms are magnetically equivalent which is in good agreement with reported analogs.17 The formation of 4 was further confirmed by LIFDI mass spectrometry. It displays the molecular ion in the mass spectrum at m/z 1176.6, corresponding to the formation of a silver cation 4, (Fig. S11, ESI†). Compound 4 has a melting point in the range of 248–255 °C as determined by differential scanning calorimetry.
In conclusion, we report on the Au and Ag complexes of amidinate-supported divalent silicon and phosphane that are capable of forming a gold-containing six-membered ring complex (2). Compound 2 exhibits intramolecular aurophilic interaction, which is further supported by quantum chemical calculations. Moreover, ligand 1 also stabilized the silver cation with two phosphane groups, giving complex 4. Noteworthy, the formation of 4 happens when the silylene moieties are oxidized by the fluorine atoms of AgSbF6 counterion, with simultaneous reduction of Sb(V) to Sb(0) as black precipitate and formation of the AlCl4− counterion.
D. S. acknowledges partial funding from the Danish National Research Foundation (DNRF93) Center of Materials Crystallography. C. M. L. thanks the Fonds der Chemischen Industrie for financial support for her PhD studies. L. Zhao acknowledges the financial support from the National Natural Science Foundation of China (Grant No. 21973044), Natural Science Foundation of Jiangsu Province (Grant No. BK20211587), the “Jiangsu Specially-Appointed Professor Plan” and Nanjing Tech University (Grant No. 39837123 and 39837132). We also appreciated the high-performance center of Nanjing Tech University for supporting the computational resources.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C.-W. So, H. W. Roesky, J. Magull and R. B. Ostwald, Angew. Chem., Int. Ed., 2006, 45, 3948–3950 CrossRef CAS PubMed; (b) S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123–1126 CrossRef CAS PubMed.
- S. S. Sen, J. Hey, R. Herbst-Irmer, H. W. Roesky and D. Stalke, J. Am. Chem. Soc., 2011, 133, 12311–12316 CrossRef CAS PubMed.
- (a) M. Haaf, R. Hayashi and R. West, J. Chem. Soc., Chem. Commun., 1994, 33–34 Search PubMed; (b) A. Fürstner, H. Krause and C. W. Lehmann, Chem. Commun., 2001, 2372–2373 RSC; (c) A. G. Avent, B. Gehrhus, P. B. Hitchcock, M. F. Lappert and H. Maciejewski, J. Organomet. Chem., 2003, 686, 321–331 CrossRef CAS; (d) W. Wang, S. Inoue, S. Yao and M. Driess, J. Am. Chem. Soc., 2010, 132, 15890–15892 CrossRef CAS PubMed.
- (a) B. Blom, S. Enthaler, S. Inoue, E. Irran and M. Driess, J. Am. Chem. Soc., 2013, 135, 6703–6713 CrossRef CAS PubMed; (b) W. Wang, S. Inoue, S. Enthaler and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 6167–6171 CrossRef CAS PubMed; (c) W. Wang, S. Inoue, E. Irran and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 3691–3694 CrossRef CAS PubMed; (d) B. Blom, D. Gallego and M. Driess, Inorg. Chem. Front., 2014, 1, 134–148 RSC; (e) D. Gallego, S. Inoue, B. Blom and M. Driess, Organometallics, 2014, 33, 6885–6897 CrossRef CAS; (f) G. Tan, S. Enthaler, S. Inoue, B. Blom and M. Driess, Angew. Chem., Int. Ed., 2015, 54, 2214–2218 CrossRef CAS; (g) R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40, 712–719 CrossRef CAS PubMed.
- (a) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC; (b) P. Pyykkö, Chem. Rev., 1997, 97, 597–636 CrossRef PubMed; (c) P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed; (d) V. W.-W. Yam and E. C.-C. Cheng, Chem. Soc. Rev., 2008, 37, 1806–1813 RSC; (e) N. Biricik, Z. Fei, R. Scopelliti and P. J. Dyson, Eur. J. Inorg. Chem., 2004, 4232–4236 CrossRef CAS For aurophilic interactions and emission properties of gold complexes, see; (f) M. M. Ghimire, V. N. Nesterov and M. A. Omary, Inorg. Chem., 2017, 56, 12086–12089 CrossRef CAS PubMed; (g) J. C. Vickery, M. M. Olmstead, E. Y. Fung and A. L. Balch, Angew. Chem., Int. Ed. Engl., 1997, 36, 1179–1181 CrossRef CAS; (h) J.-H. Jia and Q.-M. Wang, J. Am. Chem. Soc., 2009, 131, 16634–16635 CrossRef CAS PubMed; (i) R. L. White-Morris, M. M. Olmstead, F. Jiang, D. S. Tinti and A. L. Balch, J. Am. Chem. Soc., 2002, 124, 2327–2336 CrossRef CAS PubMed; (j) P. G. Jones, Gold Bull., 1981, 14, 102–118 CrossRef CAS; (k) W. Lu, N. Zhu and C. M. Che, J. Am. Chem. Soc., 2003, 125, 16081–16088 CrossRef CAS PubMed; (l) L. Rodríguez, M. Ferrer, R. Crehuet, J. Anglada and J. C. Lima, Inorg. Chem., 2012, 51, 7636–7641 CrossRef PubMed; (m) S. H. Lim, J. C. Schmitt, J. Shearer, J. Jia, M. M. Olmstead, J. C. Fettinger and A. L. Balch, Inorg. Chem., 2013, 52, 823–831 CrossRef CAS PubMed; (n) R. L. White-Morris, M. M. Olmstead, A. L. Balch, O. Elbjeirami and M. A. Omary, Inorg. Chem., 2003, 42, 6741–6748 CrossRef CAS PubMed; (o) T. K.-M. Lee, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 17646–17648 CrossRef CAS PubMed; (p) M. Saitoh, A. L. Balch, J. Yuasa and T. Kawai, Inorg. Chem., 2010, 49, 7129–7134 CrossRef CAS; (q) D. Rios, D. M. Pham, J. C. Fettinger, M. M. Olmstead and A. L. Balch, Inorg. Chem., 2008, 47, 3442–3451 CrossRef CAS PubMed; (r) H. Schmidbaur, Gold Bull., 2000, 33, 3–10 CrossRef CAS; (s) S. Pal, N. Kathewad, R. Pant and S. Khan, Inorg. Chem., 2015, 54, 10172–10183S CrossRef CAS PubMed; (t) S. Khan, S. K. Ahirwar, S. Pal, N. Parvin and N. Kathewad, Organometallics, 2015, 34, 5401–5406 CrossRef CAS.
- (a) C. I. Someya, M. Haberberger, W. Wang, S. Enthaler and S. Inoue, Chem. Lett., 2013, 42, 286–288 CrossRef CAS; (b) D. Gallego, A. Brück, E. Irran, F. Meier, M. Kaupp, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 15617–15626 CrossRef CAS PubMed; (c) M. Stoelzel, C. Praesang, B. Blom and M. Driess, Aust. J. Chem., 2013, 66, 1163–1170 CrossRef CAS; (d) A. Brück, D. Gallego, W. Wang, E. Irran, M. Driess and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 11478–11482 CrossRef PubMed.
- C. Boehme and G. Frenking, Organometallics, 1998, 17, 5801–5809 CrossRef CAS.
- P. Jutzi and A. Möhrke, Angew. Chem., Int. Ed. Engl., 1990, 29, 893–894 CrossRef.
- M. Theil, P. Jutzi, B. Neumann, A. Stammler and H.-G. Stammler, J. Organomet. Chem., 2002, 662, 34–42 CrossRef CAS.
- (a) S. Khan, S. Pal, N. Kathewad, I. Purushothaman, S. Deb and P. Parameswaran, Chem. Commun., 2016, 52, 3880–3882 RSC; (b) R. Bianchi, G. Gervasio and D. Marabello, Inorg. Chem., 2000, 39, 2360–2366 CrossRef CAS PubMed; (c) U. Flierler, M. Burzler, D. Leusser, J. Henn, H. Ott, H. Braunschweig and D. Stalke, Angew. Chem., Int. Ed., 2008, 47, 4321–4325 CrossRef CAS PubMed.
- M. Nazish, M. M. Siddiqui, S. K. Sarkar, A. Münch, C. M. Legendre, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Chem. – Eur. J., 2021, 27, 1744–1752 CrossRef CAS PubMed.
- C. Effendy, C. di Nicola, M. Fianchini, C. Pettinari, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2004, 357, 1523–1537 CrossRef.
- (a) M. Sawamura, H. Hamashima and Y. Ito, J. Org. Chem., 1990, 55, 5935–5936 CrossRef CAS; (b) S. J. Berners-Price, R. K. Johnson, A. J. Giovenella, L. F. Faucette, C. K. Mirabelli and P. J. Sadler, J. Inorg. Biochem., 1988, 33, 285–295 CrossRef CAS PubMed; (c) C. S. W. Harker and E. R. T. Tiekink, J. Coord. Chem., 1990, 21, 287–293 CrossRef CAS.
- P. Gualco, A. Amgoune, K. Miqueu, S. Ladeira and D. Bourissou, J. Am. Chem. Soc., 2011, 133, 4257–4259 CrossRef CAS PubMed.
- (a) Bruker AXS Inc., in Bruker Apex CCD, SAINT v8.30C, ed.: Bruker AXS Inst. Inc., WI, USA, Madison, 2013 Search PubMed; (b) L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed; (c) M. Sevvana, M. Ruf, I. Usón, G. M. Sheldrick and R. Herbst-Irmer, Acta Crystallogr., 2019, D75, 1040–1050 Search PubMed; (d) G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3–8 Search PubMed; (e) G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 Search PubMed; (f) C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- (a) I. Kalikhman, O. Girshberg, L. Lameyer, D. Stalke and D. Kost, J. Am. Chem. Soc., 2001, 123, 4709–4716 CrossRef CAS PubMed; (b) R. S. Ghadwal, K. Pröpper, B. Dittrich, P. G. Jones and H. W. Roesky, Inorg. Chem., 2011, 50, 358–364 CrossRef CAS PubMed.
- D. Kost, B. Gostevskii, N. Kocher, D. Stalke and I. Kalikhman, Angew. Chem., Int. Ed., 2003, 42, 1023–1026 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2125569 and 2125570. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04163d |
| This journal is © The Royal Society of Chemistry 2022 |