
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProton transfer mediated recognition of amines by ionizable macrocyclic receptors
Giuseppe
Gattuso
 *,
Daniele
Crisafulli
,
Marco
Milone
,
Francesca
Mancuso
,
Ilenia
Pisagatti
,
Anna
Notti
and
Melchiorre F.
Parisi
*
*,
Daniele
Crisafulli
,
Marco
Milone
,
Francesca
Mancuso
,
Ilenia
Pisagatti
,
Anna
Notti
and
Melchiorre F.
Parisi
*
Dipartimento di Scienze Chimiche, Biologiche, Farmaceutiche ed Ambientali, Università degli Studi di Messina, Viale F. Stagno d’Alcontres, 31, 98166 Messina, Italy. E-mail: ggattuso@unime.it; mparisi@unime.it
First published on 22nd August 2022
Abstract
Ammonium ion/carboxylate ion pairing is a key interaction ubiquitous in biological systems, but amine recognition by ionizable molecular receptors, mediated by host-to-guest proton transfer, has too often been overlooked as a design element for molecular recognition. This survey will show that proton transfer mediated recognition is a powerful and versatile tool that can be made to work with different amines and diverse macrocyclic scaffolds, such as crown ethers, calixarenes or pillararenes. We will trace the history of this recognition motif since Cram's first report half a century ago down to the latest applications in supramolecular sensing, drug-delivery and materials science, highlighting along the way the impact of host-to-guest proton transfer on self-assembly and molecular recognition.
 Giuseppe Gattuso | Giuseppe Gattuso received his PhD (1997) under the supervision of Nobel Laureate Professor Sir J. Fraser Stoddart at Birmingham University (UK). He joined the Università di Messina, where he was promoted to Assistant Professor in 1999, Associate Professor in 2014, and Full Professor in 2021. His current research interests focus on the design and synthesis of macrocyclic compounds and their use as sensors and as building blocks for the self-assembly of ordered supramolecular aggregates (1D and 2D supramolecular polymers), and on the isolation and characterization of natural compounds with bioactive potential from plant sources. |
 Daniele Crisafulli | Daniele Crisafulli obtained his bachelor's degree in 2018 and his master's degree in 2021 at the Università di Messina. Since October 2021, he has been a PhD student under the supervision of Professor G. Gattuso. He is carrying out research on the host–guest properties of pillararenes and related aromatic macrocycles and their use in sensing applications. |
 Marco Milone | Marco Milone graduated in Chemistry in 2018 at Università di Messina where he subsequently received his PhD in Chemical Sciences (2022), under the supervision of Professor Parisi. His doctoral thesis focused on the synthesis of tubular-shaped macrocyclic derivatives and the study of their supramolecular behaviour. |
 Francesca Mancuso | Francesca Mancuso received her PhD degree (2020) from the Università di Messina, working under the supervision of Professor R. Gitto. In 2020, she joined the research group of Professor G. Gattuso as a Research Fellow, carrying out research on the development of pillararene-based self-diagnostic polymers. |
 Ilenia Pisagatti | Ilenia Pisagatti obtained her PhD in Chemistry from Università di Messina in 2003, under the supervision of Prof. M. F. Parisi. After six years as Post-Doc, she spent two years as a process chemist at the academic spin-off ANMR srl. She rejoined Parisi's group in 2012 as a Research Fellow and since 2015 she has been holding the position of a technologist at the Università di Messina. She received the national habilitation to the position of Associate Professor in 2021. Her research interest focuses on the use of calixarenes and pillararenes for the construction of supramolecular polymers and drug delivery systems. |
 Anna Notti | Anna Notti received her PhD in Chemistry in 1999 from the Università di Messina. She was appointed as Assistant Professor in 2000, and Associate Professor in 2014 at the same University. Her research activity concerns the synthesis of water-soluble macrocycles (calixarenes and pillararenes) to be used as molecular receptors, sensors, multicomponent architectures, and in the solubilization and delivery of bioactive molecules. |
 Melchiorre F. Parisi | Melchiorre F. Parisi studied Chemistry at the Università di Messina, received his PhD from Oxford University in 1986, under the supervision of Professor Sir Jack Edward Baldwin, and subsequently joined Professor Robert Heinz Abeles’ group at Brandeis University as a CNR-NATO Postdoctoral Fellow (1987–1988). He currently holds the position of Full Professor of Organic Chemistry at the Università di Messina where he previously served as a ‘Ricercatore’ and then as Associate Professor (2000–2010). His current research interests lie in the field of organic synthesis related to the host–guest and supramolecular chemistry of calixarene and pillararene systems. |
Introduction
Proton transfer reactions are possibly the most general processes among the countless transformations that take place in the chemical space, since the translation of a nucleus devoid of electrons from a donor site – a Brønsted–Lowry acid – to an acceptor site – a base – can proceed without significant perturbation of the bonding electrons of the neighbouring heavier atoms involved. This elementary reaction is the foundation of an uncountable number of chemical, physical and biological processes, being pivotal especially – but not only – to all those events that take place in water or in protic solvents. However, proton transfer reactions take place in all kinds of environment, spanning from protic media to dipolar aprotic or apolar aprotic solvents, or even in the solvent-free gas phase or in the solid state. Thus, the knowledge about the acidity and basicity of organic and inorganic species is essential to exert control over the rate of a reaction or to predict its outcome.Solvents have a profound influence on the proton transfer reaction, affecting both the rate and the equilibrium position of the dissociation reaction. In fact, the acidity measured in a given medium is the ultimate combination of the intrinsic acidity (i.e., the gas phase acidity, defined as the Gibbs free energy of the acid dissociation reaction)1–3 of a HA proton donor with many different effects determined by the interactions between the solvent and the solute, such as electrostatic, hydrogen bonding, dipolar and dispersion interactions. Most of the investigations have been carried out in water or in protic solvents, along with studies in sufficiently acidic/basic dipolar non protic ones, and the values obtained are affected by solvation and by the solvent's own acidity. The acidity measured in apolar non protic media, on the other hand, is relatively less solvent dependent, and the proton transfer reaction displays a simpler, more direct mechanism.4,5 Proton transfer between an acid, HA, and a base, B, may lead to two different scenarios, a hydrogen-bonded species (AH⋯B) or an ion pair (A− BH+), as described in eqn (1)
| HA + B ⇄ AH⋯B ⇄ A− BH+ | (1) |
Such a different outcome depends on several factors, one of them being the energy needed to generate charge separation from two neutral molecules. In protic or dipolar solvents, ion solvation provides sufficient energy to drive the proton transfer. In apolar solvents, on the other hand, there are no favourable interactions to stabilize ionized solutes, so hydrogen-bonded complexes will be preferably formed.
For proton transfer to take place, energy must be found elsewhere.6 Addition to the acid–base system of a receptor capable of recognizing one (or both) ion(s) may provide, by means of a concert of weak interactions, the energy needed to tip the scale in favour of ionization. Multipoint molecular recognition may thus replace ion-solvating molecule interactions, allowing charges to be distributed from the ionic substrates to the receptor counterpart.
The influence of a macrocyclic receptor on the position of the equilibria in eqn (1) may be cleverly exploited by translating these concepts to host–guest chemistry. Rather than dealing with a three-component system where the receptor is used as an agent to influence the acid/base proton transfer, it may be envisaged that the incorporation of the acidic function in the receptor may allow applying the principles described above to the interaction between an ionizable acidic host and an ionizable basic guest (Fig. 1). As a consequence, receptor-assisted ionization may be used to provide additional stabilization to the host–guest complex by taking advantage of the electrostatic attraction of the ion pair.
 | ||
| Fig. 1 Cartoon representation of proton transfer mediated amine recognition by an ionizable macrocyclic receptor. | ||
Ion pairing – triggered by proton transfer – as a design element in host–guest chemistry has seldom been used,7 and this is surprising, given how heavily Nature relies on electrostatic interactions. For instance, ion pairing between the side-chain carboxylates of Asp or Glu and the nitrogen-containing basic moieties of Arg, Lys, or His is one of the type of bonds determining the tertiary structure of proteins. An ion pair is defined as a salt bridge if the centroids of the oppositely charged groups lie within a distance of 4.0 Å (Fig. 2).8,9 Salt bridges contribute to protein conformational stability to a different extent. A surface-exposed salt bridge may provide 0–2 kcal mol−1, whereas a buried one can be worth 3 kcal mol−1 or more.10–12 This occurs because those on the surface are involved in multiple interactions with solvating water molecules, whereas the buried ones are surrounded by a hydrophobic environment. In general, when complementary polar and charged groups are buried, they make favourable electrostatic interactions (i.e., hydrogen bonds or salt bridges) and are thus said to be “compensated”, whereas unsatisfied charges have a destabilizing effect.13
 | ||
| Fig. 2 Salt bridge existing in the carbonic anhydrase enzyme (PDB code: 3UCJ, https://www.rcsb.org/) involving the carboxylate of aspartic acid at position 49 and the guanidinium of arginine at position 51. | ||
Salt bridging within the cavity of a synthetic receptor shows some analogies to buried salt bridges, with the oppositely charged species shielded from the medium by the macrocycle walls, thus allowing for a direct, unmediated electrostatic interaction and favouring host-to-guest proton transfer (and ionization) as opposed to hydrogen bonding between non-ionized species.
In a broader perspective, much effort has been devoted to using ionized (often carboxylated) macrocycles as counterions of metal cations to improve the efficiency in liquid–liquid extraction from an aqueous to an organic phase by what basically amounts to an ion-exchange mechanism.14 In this review, we will trace the history of proton transfer-mediated recognition of amines, starting from the first case reported in the literature over 45 years ago.15 We will specifically focus on macrocyclic species bearing carboxylic acids or phenolic groups as proton-donating agents, mainly because of the ability of these functional groups to provide host-to-guest stoichiometric proton transfer in the presence of amines and form tight carboxylate–ammonium or phenoxide–ammonium ion pairs, but also for the allure of the nature-mimicking carboxylate–ammonium salt bridge interaction. The survey will be organized by macrocyclic classes, starting from crown ethers, moving to calixarenes and then down to the newest family of pillararenes.
Crown ethers
Many new ideas, concepts or applications in the field of supramolecular chemistry came along with crown ethers.16 This is certainly true for the proton transfer-mediated complexation of amines, which was first observed by D. J. Cram in 1975.15 In a paper dealing with the effect of functional groups on the binding properties of m-xylyl-containing crown ethers, the authors reported that, upon mixing carboxyl crown ether 1 with tert-butylamine in cyclohexane/dichloromethane, a crystalline one-to-one salt was formed. No special emphasis was put over this finding as the concept of proton transfer-triggered recognition was not formulated until later. It should be underlined though that an amine was directly used with carboxylic acid-bearing 1, rather than tert-butylammonium thiocyanate, that was used in that same paper to make classical charged complexes with non-ionizable crown ethers. In addition, a D2O/CDCl3 distribution experiment on 1−⊃t-BuNH3+ and the corresponding tert-butylamine complexes formed in the presence of smaller (2·H and 3·H) and larger (4·H) crown ethers showed the former being the most lipophilic supramolecular salt. Unsurprising in hindsight, as the macrocyclic cavity of 1·H has roughly the size of 18-crown-6, the better fit helps the shielding of the charged moieties from the low polarity environment.17 The solid-state structure of this complex was reported later (Fig. 3).18
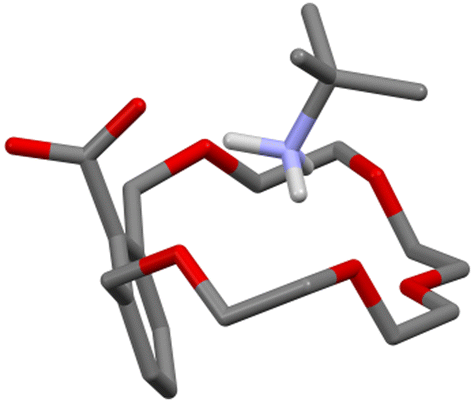 | ||
| Fig. 3 Solid-state structure of the 1−⊃t-BuNH3+ complex.18 | ||
Shortly after, McKervey et al. described two similar derivatives, 5·H and 6·H, bearing, on the xylyl ring, a phenolic hydroxyl as the proton-ionizable group.19 Again, upon exposure to ammonia or simple primary amines, proton transfer followed by recognition took place, leading to the formation of the relative complexes. UV-vis analysis indicated the complete conversion of 5·H to 5−⊃t-BuNH3+, whereas the conversion of the smaller-sized 6·H to the corresponding ion-paired complex barely reached 5%. The authors explained this behaviour by invoking the ability of 5− to stabilize NH4+ through “solvation by the crown oxygen atoms”.19
Crown 5− adducts with ammonia and methylamine, however, were found to be unstable and slowly reverted to their starting components. Conversely, nitrophenoxide derivative 7− gave stable crystalline complexes with ammonia, tert-butylamine and α-methylbenzylamine, as a consequence of the lower pKa of its conjugated acid (7·H) with respect to 5·H (6.6 vs. 10.6). Further investigations on the 7−⊃NH4+ complex revealed that in the solid state the bond lengths of the aromatic unit are consistent with the presence of an anionic p-nitrophenoxyde moiety with a significant quinoid character (Fig. 4),20 explaining in the end the higher stability of the 7− complexes.
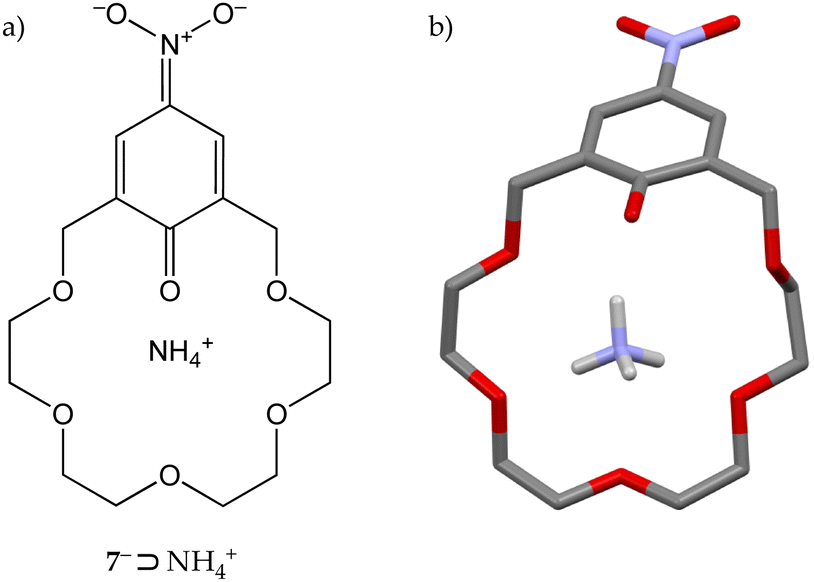 | ||
| Fig. 4 (a) Quinoid structure of the 7−⊃NH4+ complex. (b) Solid-state structure of 7−⊃NH4+.20 | ||
Host 7·H was also used for the formation of the first 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ion-paired complexes, using 1,2-diaminoethane and 1,3-diaminopropane as basic guests.20
:1 ion-paired complexes, using 1,2-diaminoethane and 1,3-diaminopropane as basic guests.20
It may be useful at this stage to comment on the profound effect played by a macrocyclic receptor on the pKa of a given Brønsted–Lowry acid moiety embedded into its framework. The acidity, as mentioned above, is a combination of many effects and, in oxygen-containing macrocycles, it may depend also on the ability of the crown ether oxygen atoms to undergo intramolecular hydrogen bonding. Cram and co-workers showed that, by going from 2,6-bis(methoxymethyl)benzoic acid to 1·H and finally to pyridine-containing crown ether 9·H (Fig. 5a), the pKa value increases from 3.3 to 4.8 and then to 5.8, respectively.21 This is the result of the increasing ability of a polyether chain (1·H) and a polyether moiety together with a pyridine ring (9·H) to hydrogen bond, and therefore ‘complex’ the carboxylic hydrogen atom, making it less likely to dissociate. Overall, intramolecular hydrogen bonding in this series is worth 2.5 pKa units. In the case of 9·H, a strong hydrogen bond was demonstrated also in the solid state, with the carboxyl proton located very close to the pyridine nitrogen (1.8 Å, Fig. 5b).21
 | ||
| Fig. 5 (a) Host 9·H and (b) its solid-state structure. Reproduced with modifications from ref. 21 with permission from the American Chemical Society, copyright 1982. | ||
The Brønsted–Lowry acid does not need to be a carboxylic or a phenolic moiety. The hydrogen atom that is transferred to the guest may also come from a nitrogen heterocycle,22,23 as shown by Izatt and co-workers in 1985.24 They synthesized a series of 1,2,4-triazole-containing crown ethers (10·H–12·H), and used them in recognition experiments with ammonium ions and amines (Fig. 6). Compound 10·H was tested against benzylamine and benzylammonium perchlorate. Both were recognized, but the free energy of activation (ΔG‡)25 was 3.6 kcal mol−1 in favour of 10−⊃BnNH3+ over 10·H⊃BnNH3+ (14.0 vs. 10.4 kcal mol−1, respectively), demonstrating that the internally ion-paired complex was kinetically more stable than the positively charged one. Based on this evidence, they synthesized a chiral triazole-containing macrocycle, (S,S)-12·H, intended for the chiral discrimination of 1-(1-naphthyl)ethylamine (NapEtNH2) enantiomers, but the experiment had a limited success. The macrocycle did recognize both enantiomeric amines, but no chiral discrimination was observed. An interesting feature, nonetheless, is that the solid-state structure of (S,S)-12−⊃(R)-NapEtNH3+ showed the triazole hydrogen atom located on the guest nitrogen atom, confirming, as programmed, a successful host-to-guest proton transfer. The authors also remarked that this was the first example of a complex composed of an alkylammonium cation and a macrocyclic receptor in which the anion is actually a part of the macrocyclic ring.24
 | ||
| Fig. 6 (a) Hosts 10·H–12·H and (b) solid-state structure of the (S,S)-12−⊃(R)-NapEtNH3+ complex.24 | ||
A natural development of these early ionizable receptors was brought forth by Kaneda and co-workers,26,27 who introduced chromogenic units to the parent scaffolds in order to trigger a macroscopic response upon amine recognition. Azophenol-containing crown ethers 13·H–17·H were then synthesized, with the aim of exploiting the transfer of the acidic phenolic proton to induce a colour change.28–30 This setup allowed the authors to explore different features of the recognition process, such as the number of ionizable sites, the size of the crown ether macrocycle and the chirality of the receptor.
Monoprotic crown ethers 13a·H–13e·H were tested against 11 simple amines, and the UV-vis spectra of the resulting mixtures were recorded.29 Data analysis revealed some interesting patterns and allowed them to draw useful conclusions: (i) the significant blue shifts of the absorption maxima observed in the case of 13b·H–13d·H (i.e., the derivatives with cavities roughly isosteric with 15-crown-5, 18-crown-6 and 21-crown-7) ruled out a simple acid–base reaction, providing, instead, strong evidence in favour of a process in which the recognition of the cationic guest is necessary to push the equilibrium towards the ionized forms; (ii) 13c·H, with its 18C6-like cavity, easily reacted with most of the amines tested, and (iii) with the exception of 13a·H, all the other derivatives positively recognized secondary amines. In particular, 13b·H recognized piperidine quite efficiently (pKa = 3.26) as confirmed by the solid-state structure of the ion-paired complex, held tightly together by two +N–H⋯O−, one +N–H⋯O and three +N⋯O interactions (Fig. 7a).29
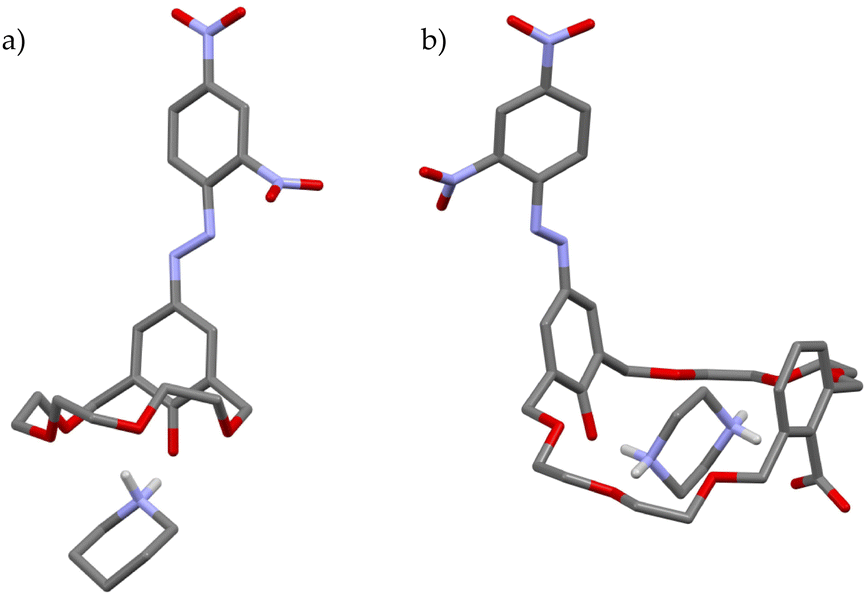
As a part of these investigations, the same authors equipped a 24-membered azophenolic crown ether with an additional ionizable site, namely a carboxyl group, reasoning that it might allow the efficient recognition of dibasic guests.28 In fact, 14·2H turned out to be moderately responsive to the presence of monoamines (primary, secondary, and tertiary alkylamines, piperidines and ethanolamines), showing absorption maxima in the 580–590 nm range. Conversely, when exposed to diamines such as hydrazine, piperazine, N,N′-dimethyl-piperazine, 1,4-diazabicyclo[2.2.2]octane, or N,N,N′,N′-tetramethylethylenediamine, salt formation displayed intensely blue-shifted maxima in the 535–549 nm range. In particular, single crystals of the 1:1 complex of 14·2H with piperazine provided concrete evidence in favour of a double proton transfer process (Fig. 7b). This study is an elegant demonstration of the importance of the steric- and stereo-electronic matching between the host and guest: although proton transfer to monoamines does take place, double proton transfer to a single diamine is very efficient (Ka = 2.3 × 106 M−1 for the 1:1 piperazine complex). In fact, the addition of a second piperazine molecule to make a 1:2 complex proceeded less smoothly, with an estimated Ka for this second binding event smaller by three orders of magnitude (3.3 × 103 M−1). Based on these findings, they concluded that the extent of blue-shifts is likely associated with the formation of more stable salts, suggesting that, in the case of piperazine, hydrogen bonding between the phenolate oxygen and an ammonium hydrogen stabilizes the energy of the polar ground state more than that of the less polar excited state of the chromophore.28
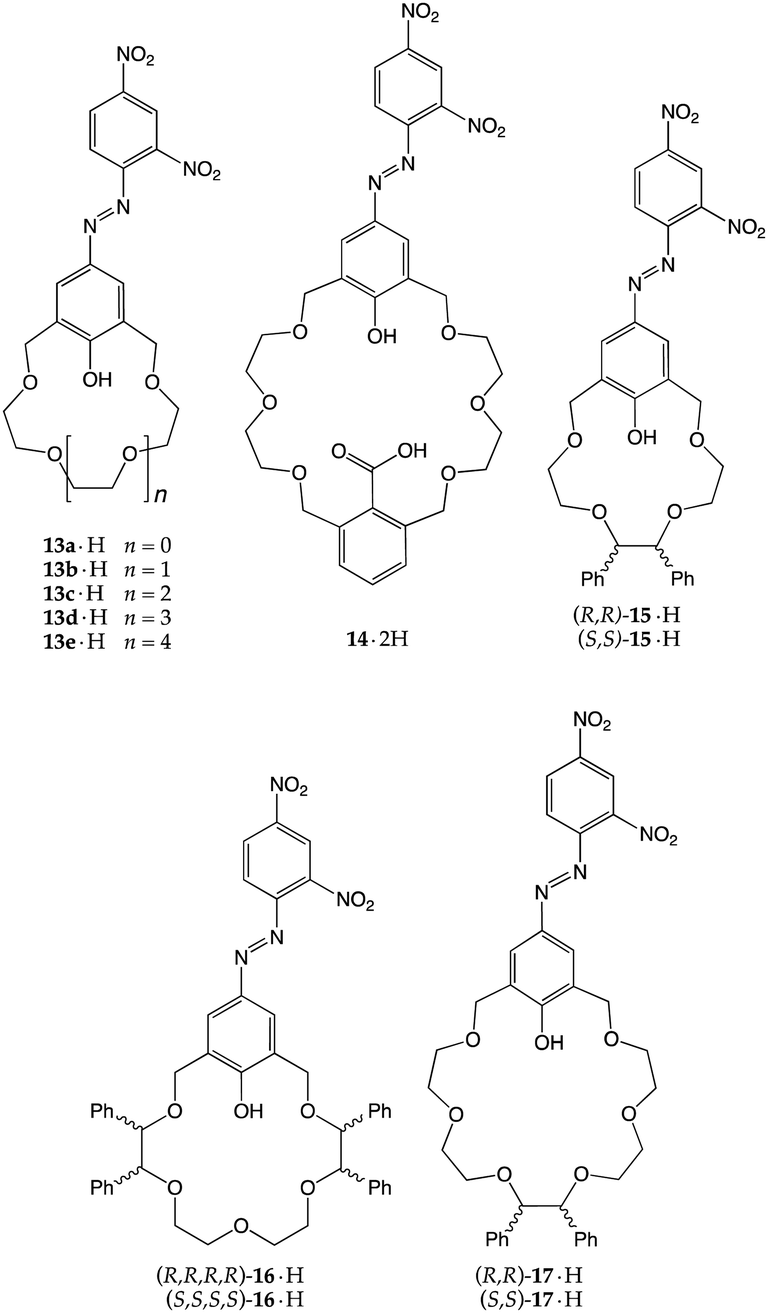
Chiral azophenolic crown ethers were also reported for the enantioselective recognition of chiral amines.30 Three enantiomeric pairs of receptors were prepared (15·2H–17·2H, with 15-, 18- and 21-membered rings, respectively) and assayed against thirteen chiral primary alkyl- and ethanolamines. Differences (up to 11 nm) in λmax between diastereomeric sets of crown–amine complexes were observed, indicating that the proton transfer-mediated recognition was taking place with some enantiomeric discrimination. The authors also attempted to predict the absolute stereochemistry by a clever analysis of the potential ammonium binding modes (two- or three-point) conducted on the CPK models of the complexes, speculating that the host–guest pairs for which better complementarity was predicted were those which exhibited the larger blue shift.
The naked-eye colorimetric detection of amines can serve more sophisticated purposes, namely, the determination of the chain length of α,ω-diamines by making use of the suitable phenolphthalein bis-crown ether 18·2H.31 Phenolphthalein develops a colour when is present in its dianionic form, after the opening of the spirolactone ring. Upon addition of linear α,ω-diamines (H2N(CH2)nNH2, n = 4–10), an intense violet coloration was observed for the derivatives with 8 and 9 carbon atoms, whereas a timid response was seen for the 7- and 10-membered guests (Fig. 8).
 | ||
| Fig. 8 (a) UV-visible spectra of methanol solutions of 18·2H in the presence of different diamines (H2N(CH2)nNH2 (n = 4–10). (b) Colour changes of the solutions of the different complexes. Reproduced with modifications from ref. 31 with permission from the American Chemical Society, copyright 1999. | ||
No colour developed with shorter diamines or test monoamines. All considered, 18·2H recognized only those guests able to span between the two crown cavities, leaving out also those longer diamines that would not fit correctly in the cleft created by the polyether rings. However. an in-depth analysis of the interaction between 18·2H and 1,9-diaminononane showed a more complex picture. A Job plot revealed that the stoichiometry was not 1:1, but leaned toward 1:2 or 2:3 host-to-guest ratios, suggesting that one diamine molecule was bridged between the crown rings of a single host molecule and another would act as the counterion of the carboxylate of two different host molecules (Fig. 9a).31 Addition of N-ethylpiperidine to the system, acting as a proton scavenger and as the carboxylate counterion, restored a 1:1 stoichiometry (Fig. 9b).
 | ||
| Fig. 9 Proposed structures of the 2:3 (a) and 1:1 (b) complexes of 18·2H with diamines.31 | ||
Phenolphthalein bis-crown ethers were also employed for the enantioselective recognition of alanine derivatives.32 Four new derivatives 19·2H–22·2H were synthesized, bearing pairs of stereogenic centres at different positions of the polyether chains. Even though they showed a much lower affinity for diamines than the parent 18·2H, 20·2H showed the best selectivity, binding guests (R)-26 and (R)-27ca. 6 times better than the corresponding (S) enantiomers. The binding constants of the complexes between 20·2H and (S)-26 or (S)-27 were so small (62–65 M−1) that no colour developed upon the guest addition, ultimately allowing for the naked-eye discrimination of the (R)-enantiomers.
Derivative 18·2H was also employed for the recognition of dipeptides bearing lysine residues at the C-terminus.33 400 different polymer-supported dipeptides were tested, but the colour development was observed only for Gly–Lys, Ala–Lys, Ser–Lys, Thr–Lys, Arg–Lys, and Lys–Lys. By taking advantage of this selectivity, 18·2H was used for the visual recognition of proteins with a (Gly, Ala, Ser, Thr, Arg, or Lys)-Lys sequence at their N-terminus, such as the amyloid β/A4 protein precursor (APP770(394–410)) and scyliorhinin I.
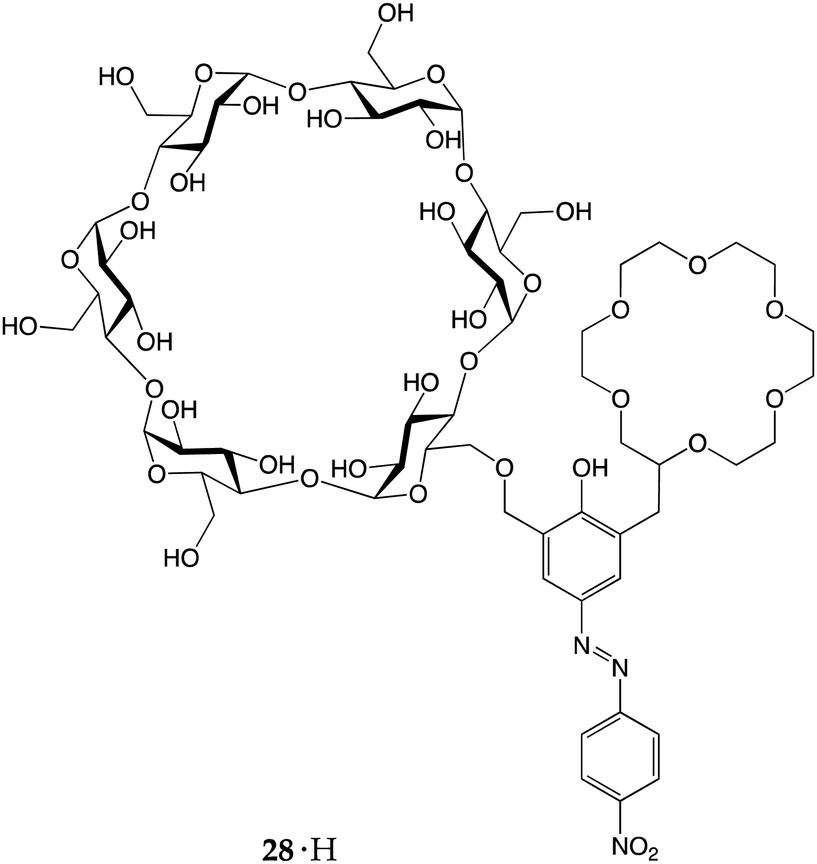
More recently, Kaneda and co-workers reported an azophenolic crown ether equipped with α-cyclodextrin as an additional binding site.34 Ditopic receptor 28·H was able to efficiently discriminate between primary, secondary and tertiary amines. In fact, upon proton transfer, different amines had different fates. The addition of primary and secondary amines to 28·H induced intense bathocromic shifts to the absorbance maximum, from 380 to ∼580 and ∼530 nm, respectively. The log Ka values of 28·H complexes with primary and secondary amines were found to be 4.19–4.85 and 2.02–2.32, respectively, whereas tertiary amines did not display any colour change. The key to the selectivity of this receptor lies in the concerted action of the crown ether and the cyclodextrin sub-units. Upon the host-to-guest proton transfer, the resulting alkylammonium ions interact with the anionic receptor in a different fashion: the primary one can accommodate its alkyl chain within the CD cavity and, is so doing, establishes three H-bonds with the polyether ring and an +N⋯O− electrostatic interaction with the azophenolate moiety; the secondary and tertiary ones, on the other hand, can fit only one of their alkyl chains (the others increasing steric hindrance) into the cavity and, because of the lower number of hydrogen atoms bound to the N+, can establish only two (or one) H-bonds with the crown ether moiety.
Calix[n]arenes
Ionizable macrocycles have been extensively investigated by the group of Bartsch, who published several papers on the use of carboxyl calix[4]arenes,35 crown ethers14 (especially lariat ethers36) and calix[4]crowns37 as metal ion extractants. They synthesized a huge number of ionophores over the years, exploring the influence of the number and the tri-dimensional arrangement of the carboxylate moieties on the extraction efficiency and selectivity of singly charged and multi-charged ions. This approach was very straight to the point, using ionophores fitted with a number of carboxylate groups able to match the charge of the target metal cation (mismatched systems fall outside the scope of this review article).38–42 However, they did not focus their attention on amines, with perhaps the only exception being a carboxyl lariat crown ether used as a host for the recognition of the terminal amino group of siderophore ferrioxamine B.43The earliest example of proton transfer-triggered recognition by calixarenes was reported by Fukazawa and co-workers, who employed a calix[5]arene scaffold to place two benzoic acid moieties facing each other at the wider rim.44 Compound 29·2H was then tested against imidazole, providing evidence of a complexation process triggered by proton transfer. Further investigation showed that the diprotic host 29·2H and monobasic imidazole formed a 1:2 host/guest complex, with one imidazolium ion likely included inside the π-rich cavity of the calixarene and the second one tweezed between the carboxylate groups.
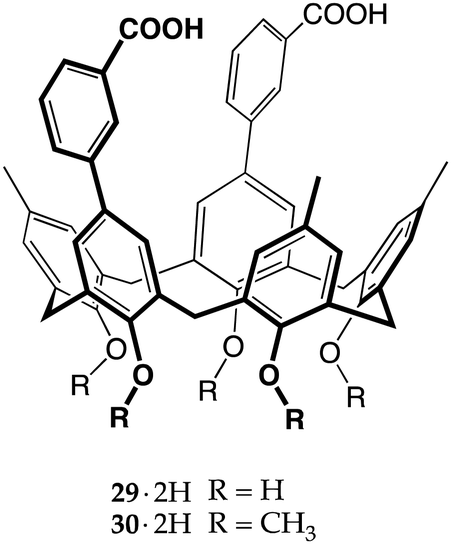
Calix[5]arene 29·2H and its penta-O-methyl derivative 30·2H were subsequently used for the recognition of guests with more than one basic nitrogen atom, such as 2-aminopyrimidine and 9-ethyladenine.45 Leaving aside the poor results obtained with 30·2H (methylation of the narrow rim significantly reduces the preorganization of the calixarene, owing to the loss of the H-bonds between the phenolic OHs),4629·2H showed an excellent affinity for 9-ethyladenine over 2-aminopyrimidine (Kas 4.3 × 104vs. 8.7 × 102 M−1, respectively). The authors insightfully explained that, in order to complex the smaller pyrimidine, 29·2H needs to bring the carboxylates close to each other, weakening (or breaking) the H-bonds at the narrow rim of the macrocycle. The adenine derivative, on the other hand, would fit perfectly in the cleft generated by the two benzoate moieties of the calix[5]arene, held tightly by both Watson–Crick and Hoogsteen type H-bond interactions.45
In 1996, Kubo and co-workers reported a calix[4]arene-crown that elegantly combined all the features discussed above (Fig. 10).47 Macrobicyclic derivative 31·2H contains two indophenolic chromophores – embedded in the calixarene scaffold – capable of performing proton transfer and, in so doing, returning an intense colorimetric response. In addition, the presence of a chiral binaphthol moiety gave receptor 31·2H the ability to discriminate enantiomers. The addition of chiral aminoalcohol, (R)-phenylglycinol, to a solution of 31·2H (λmax = 515 nm) resulted in an immediate colour change from red to blue-violet, with the appearance in the UV-vis spectrum of two intense bands at 538 and 652 nm. Conversely, no colour change was observed upon the addition of the enantiomer, (S)-phenylglycinol, nor upon exposure to the structurally related substrates, hydroxyl-lacking (R)- or (S)-1-phenylethylamine.
 | ||
| Fig. 10 (a) Calix[4]arene-crown 31·2H and (b) the solid-state structure of its complex with (R)-phenylglycinol.47 | ||
Based on their observations, the authors proposed a very detailed binding model for 31·2H and (R)-phenylglycinol (Fig. 10b). Firstly, a proton transfer from one phenolic group of the host to the amino guest has to take place, so that the resulting ammonium ion may bind to the crown ether moiety of [31·H]−. Additional stabilization is provided by an OH⋯−OAr hydrogen bond and a hydrophobic interaction between the binaphthyl unit and the aromatic ring of the guest. According to the authors, the latter interaction justifies the shorter wavelength emission observed in the UV-vis spectrum of the complex, whereas, the longer one, originates from the ionized indophenolate group. (S)-Phenylglycinol would suffer from steric hindrance from the binaphthyl rings, whereas (R)- and (S)-1-phenylethylamine would lack the OH group to hydrogen bond to the indophenolate moiety, none of them providing the naked-eye colorimetric detection.
Following the track set by this seminal paper, a homooxacalix[3]arene receptor (32·H) was reported, bearing a dye-functionalized phenol moiety able to ionize and induce a change of colour once the proton is released.48 The colour change comes from the betaine form adopted by the Reichardt dye ET1 embedded into the macrocyclic scaffold, upon the transfer of the phenolic proton to the guest.49 Homooxacalixarene 32·H was successfully employed for the naked-eye recognition of a number of primary and secondary amines with low steric hindrance (n-hexylamine, cyclohexylamine, piperidine, 2-aminoethanol and so on).48 Remarkably, in all these instances, proton transfer took place even though the resulting 32−/ammonium complexes were found to have modest association constants (Kas in the 43–135 M−1 range).
In more recent years, our research group has exploited the proton-transfer strategy for the recognition of amines (as alkylammonium ions) by calix[5]arenes.50 Inspired by the excellent fit discovered in the solid-state structure of the complex between the n-butylammonium ion and calix[5]arenes 33 and 34,51,52 we envisioned that we could take advantage of a ionizable group placed at the narrow rim of the calixarene cavity as a proton source. Carboxyl calix[5]arenes 35·H and 36·H were prepared, and tested for their affinity for n-butylamine.53 In line with our predictions, both derivatives were able to recognize n-BuNH2 very efficiently. Titration experiments showed that proton transfer took place as programmed, triggering the endo-cavity inclusion of the resulting butylammonium ion in both cases (Ka = 3.21× 104 and 1.13× 104 M−1 for 35−⊃n-BuNH3+ and 36−⊃n-BuNH3+, respectively). The solid-state structures of 35−⊃n-BuNH3+ and 36−⊃n-BuNH3+ revealed the formation of supramolecular host–guest salts (i.e., “internal” ion-paired complexes), but only in the former case the calixarene carboxylate group was salt-bridged to the n-butylammonium ion guest (Fig. 11).53

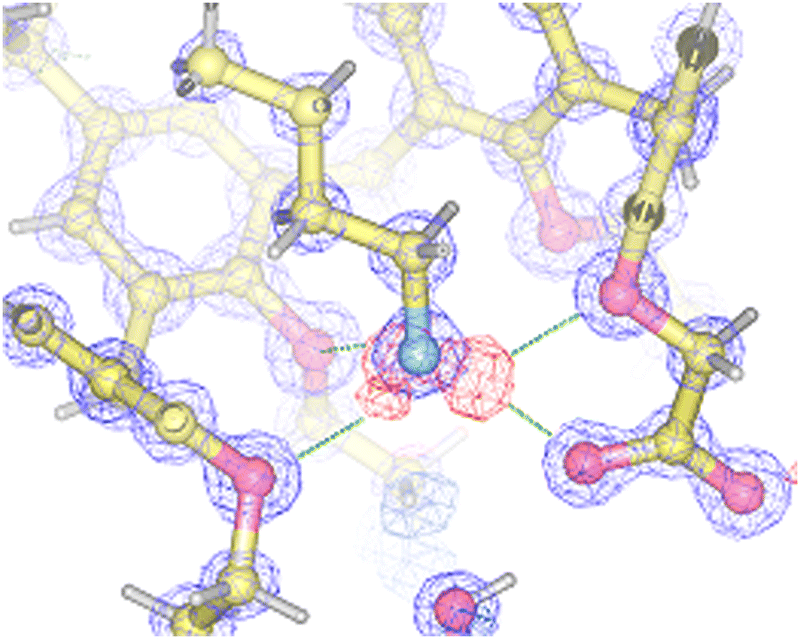 | ||
| Fig. 11 Section of the electron density map and the residual electron density map observed for the 35−⊃n-BuNH3+ complex, showing the position of the hydrogen atoms involved in H bonds. Reproduced with permission from the American Chemical Society.53 | ||
These receptors also proved to be extremely selective for primary alkylamines. When treated with n-BuNH2, n-Bu2NH and n-Bu3N, only the primary one was protonated and included within 35·H, confirming that complexation after the protonation step is mandatory to provide the energy required to generate the charged species.53,54 Furthermore, among primary alkylamines, linear ones are selectively recognized over branched ones, with 35·H being able to pick n-BuNH2 out from a mixture containing i-BuNH2, s-BuNH2 and t-BuNH2 in equal concentrations.53 This study highlights the key role played by the solvent in the proton transfer process: switching from chloroform to chloroform/methanol (9:1 v/v) and then to chloroform/trifluoroethanol (9:1 v/v), the Ka values progressively increase, owing to a more efficient solvation of the ionized species.
Calix[5]arenes are known to form capsular 2:1 complexes with linear α,ω-alkanediyldiammonium salts of suitable length.55 Ionizable calixarenes 35·H and 36·H were therefore employed in a series of solution/solid-state studies for the encapsulation of diamines,56–58 reasoning that each amino end-group could be protonated and then included inside the cavity of two distinct calixarene molecules.
Calixarene 35·H was used for an investigation on the guest length requirements to form capsules, either as ‘sealed’ capsules where the two macrocycle rims would be in touch with each other, or quasi-capsular 2:1 complexes, with the two wider rims not in van der Waals contact (Fig. 12a–c).56 The solid-state structures of the complexes with 1,10-decanediamine, 1,11-undecanediamine and 1,12-dodecanediamine (35−⊃+H3N(CH2)10NH3+⊂35−, 35−⊃+H3N(CH2)11NH3+⊂35− and 35−⊃+H3N(CH2)12NH3+⊂35−, respectively) revealed that 35·H can encapsulate up to an eleven-carbon diamine, again as the result of a combination of supramolecular interactions. A close inspection of these structures showed that the capsules are held together by 20 noncovalent interactions: two endohedral carboxylate–ammonium salt bridges, six H-bonds, eight CH–π contacts, and four van der Waals contacts between the wider rims of the macrocycles. In agreement with the ideal 70% packing coefficient postulated by Mecozzi and Rebek,59 the capsules were found to be tightly packed, with about 67–72% of their internal volume occupied by the guest.
 | ||
| Fig. 12 Solid-state structures of: (a) 35−⊃+H3N(CH2)10NH3+⊂35−, (b) 35−⊃+H3N(CH2)11NH3+⊂35−, (c) 35−⊃+H3N(CH2)12NH3+⊂35−, (d) 36−⊃+H3N(CH2)10NH3+⊂36−, (e) 36−⊃+H3N(CH2)11NH3+⊂36−, and (f) 36−⊃+H3N(CH2)12NH3+⊂36−.56,57 | ||
Calixarene 36·H showed a different picture in the presence of the same diamino guests (Fig. 12d–f).57 2:1 Capsular or quasi-capsular complexes did form as a result of a proton transfer driven molecular recognition process; a surprising feature, however, was the orientation of the carboxylate ions in the solid-state structures. Rather than lean inward to form salt bridges with the ammonium ions, similarly to what was seen for 35−⊃n-BuNH3+, they pointed away from the cavity, preferring to H-bond with CF3CH2OH crystallization molecules. In addition, only 35−⊃+H3N(CH2)10NH3+⊂35− appeared as a sealed capsule.
Moreover, a comparison between the capsules and the quasi-capsules formed in the presence of 35·H and 36·H highlighted a peculiar arrangement of the calixarene units with respect to each other. Looking along the guest axis they adopt an eclipsed arrangement in the capsules and a staggered one in the quasi-capsules. In other words, the facing calixarenes ‘rotate’ with respect to each other to seal the inner capsular space with a kind of hydrophobic (van der Waals) ‘bayonet-mount’ fastening (Fig. 13).57
 | ||
| Fig. 13 Schematic representation of the relative orientation of the two calix[5]arene units in the solid-state structure of the capsular and quasi-capsular complexes. The red and blue spheres represent the phenolic oxygen atoms. (a) 35−⊃+H3N(CH2)10NH3+⊂35−, (b) 35−⊃+H3N(CH2)11NH3+⊂35−, (c) 35−⊃+H3N(CH2)12NH3+⊂35−, (d) 36−⊃+H3N(CH2)10NH3+⊂36−, (e) 36−⊃+H3N(CH2)11NH3+⊂36−, and (f) 36−⊃+H3N(CH2)12NH3+⊂36−.57 | ||
In a subsequent paper, this design based on the proton transfer-mediated recognition of diamines followed by salt bridge-assisted bis-endo-cavity inclusion was applied to the sequestration of the biogenic amines, spermine and norspermine.58 Remarkably, upon exposure to these tetrabasic guests, the two calixarene units of 35·H involved in the encapsulation transferred their protons only – and precisely – to the primary amine groups, generating a charge-matched 2:1 complex with positive charges on the sites that ensure maximum electrostatic interaction, thus demonstrating the potential and the versatility of this recognition motif.
 | ||
| Fig. 14 Synthesis of pillar[5]arene-based [1]rotaxane from carboxylic acid 38·H.67 | ||
Pillar[n]arenes
Recent reports on carboxyl pillar[5]arenes ideally connect the most recent macrocyclic molecular receptors to Cram's work on crown ethers from almost half a century ago.15 Pillar[n]arenes are a family of para-cyclophanes consisting of hydroquinone units connected by methylene bridges, possessing a π-rich prismatic cavity. Since they were first reported by Ogoshi in 2008,60 they have attracted tremendous attention, so much so that over the past fourteen years nearly 1400 papers have been published on their molecular and supramolecular properties.61–63 Proton transfer-mediated recognition was bound to cross paths with the chemistry of these new receptors, and it first did it in two papers published by Yu and co-workers in 2014.64,65They reported the synthesis of dicarboxyl pillar[5]arene 37·2H, and extensively investigated its ability to recognize α,ω-diamines of different length. Diamines are recognized by pillararenes by means of CH⋯π interactions between the alkyl chain of the guest and the aromatic walls of the host.66 The authors found that proton transfer occurs and that the resulting diammonium ions are included inside the pillararene cavity, taking advantage of the additional electrostatic interactions between the ionized groups. It is worth remarking that 1,8-octanediamine binds to the non-ionizable decamethylpillar[5]arene with a Ka of 70 M−1 (in chloroform),66 whereas it binds to 37·2H (as a di-ionized 372−⊃+H3N(CH2)11NH3+ complex) with a Ka of 2.89 × 105 M−1 (in a chloroform/methanol 1:1 v/v mixture).64
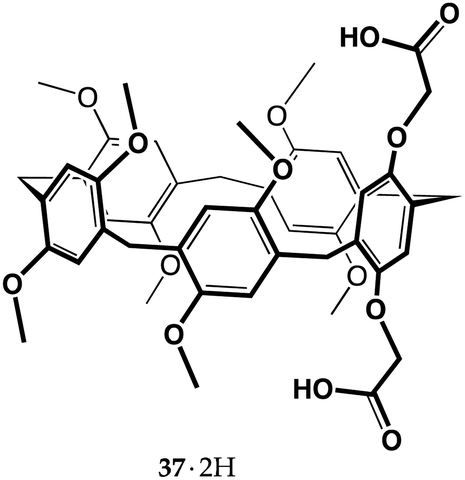
Diamines with odd and even numbers of carbon atoms displayed a peculiar trend, the former showing higher Kas than the latter, probably because in an ideal zig-zag conformation the odd-numbered alkanediyldiammonium ion is able to reach the two carboxylate groups of the host more easily, as their nitrogen atoms point in the same direction. Determination of the thermodynamic parameters by isothermal titration calorimetry (ITC) revealed that all 37·2H complexes featured ΔH° < 0, TΔS° < 0 and |ΔH°| > |TΔS°|, indicating that these recognition events were all enthalpy-driven.64,65
The papers discussed so far deal mostly with fundamental research and sensing applications. The synthesis of a pillar[5]arene-based [1]rotaxane is the first example of proton-transfer mediated recognition used as a tool to build a more complex molecular architecture.67 [1]Rotaxanes are a peculiar group of mechanically interlocked molecules (MIMs) where the wheel and the axle are also covalently linked to each other.68 Carboxyl pillar[5]arene 38·H first recognizes 10-(3,5-dimethoxyphenoxy)decaneamine, holding it tightly inside the cavity by harnessing the electrostatic attraction between the carboxylate of the host and the ammonium ion of the guest. Once formed, the complex was treated with an amide coupling agent to yield [1]rotaxane 39 in a 73% yield (Fig. 14).
An analogous approach was employed for the synthesis of the even more exotic pillararene-based MIM 40 (Fig. 15), a pseudo[1]catenane. These macrobicyclic systems, owing to their ability to self-include one macrocyclic moiety inside the cavity of the other, may somehow be likened the catenane family. Huang and co-workers combined suitable host–guest pairs (37·2H and diethylenetriamine or triethylenetetramine) and succeeded to obtain pseudo[1]catenanes 40a,b (Fig. 15).69 As in the case of calix[5]arene 36·H and spermine (see above), proton transfer takes place with extreme precision, with a double release of the carboxyl protons from 37·2H to the basic primary amino sites of the guests, to secure, upon complexation, additional carboxylate/ammonium interactions. Again, the treatment of such complexes with a coupling agent led to the desired products 40a,b.
 | ||
| Fig. 15 Synthesis of pseudo[1]catenane 40a,b from dicarboxyl pillar[5]arene 37·2H.69 | ||
These pseudo[1]catenanes turned out to have very interesting properties. Pillararenes possess planar chirality, although pS and pR enantiomers are often impossible to separate as a result of a fast interconversion (via the intra-annular rotation of the five aromatic rings) at room temperature.70 In pseudo[1]catenanes, on the other hand, self-inclusion/exclusion leads to an equilibrium between in and out conformers, generating two pairs of atropoisomers (in-pS and out-pR as well as in-pR and out-pS). Therefore, each enantiomer can undergo a conformational rearrangement into its atropoisomer and, as a result, invert its planar chirality. The control over these equilibria allows these compounds to be used as chiroptical sensors (Fig. 16).
 | ||
| Fig. 16 Inversion of the chirality of pseudo[1]catenanes 40.69 | ||
Latest developments
Very recently, more intriguing applications – sensing, drug-delivery, smart materials – have started to surface in the literature. In 2021, for instance, a pillar[5]arene dicarboxylic acid equipped with a group able to give aggregation-induced emission (AIE) was reported.71 Pillar[5]arene 41·2H was designed to recognize α,ω-diaminoalkanes (8, 10, or 12 carbon atoms) and signals their presence with a fluorescence enhancement (Fig. 17 for 1,8-diaminooctane). The AIE effect observed in this system was rationalized in terms of a restricted rotation of the quinoline-malononitrile moiety around the σ-bonds linking it to the tiophene ring, caused by the carboxylate/ammonium interaction between the host and guest.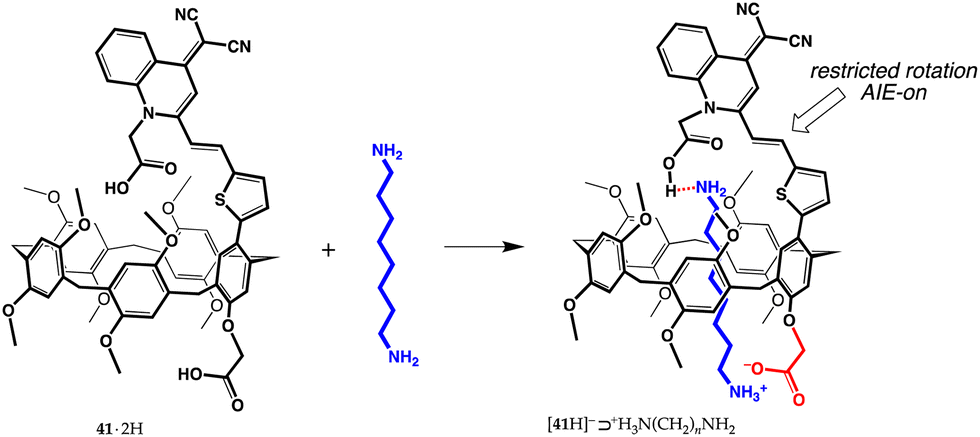 | ||
| Fig. 17 Formation of the AIE-active [41·H]−⊃+H3N(CH2)8NH2 complex.71 | ||
Pillararene 41·2H proved to be extremely sensitive, with a limit of detection, for the diamines assayed, in the 10 nM range. In silico studies provided evidence of a unique feature, a single proton transfer, from the carboxyl group directly attached to the pillararene to an amino group of the guest, with the formation of a carboxylate/ammonium salt bridge (1.447 Å). The second carboxyl group of [41·H]−⊃+H3N(CH2)8NH2, on the other hand, was seen to form an O–H⋯N charge-assisted hydrogen bond (1.682 Å), not uncommon in carboxyl/amine interactions.72
Proton transfer mediated recognition was the starting event in a 2021 report on imaging-guided drug delivery.73 A way was devised to load the anticancer drug, doxorubicin, onto nanoparticles capable of cellular internalization, and monitor the release of the cargo inside the cell nuclei. They used derivative 38·H to bind tetraphenylethylene-based AIE-gen 42 (Fig. 18). The resulting 38−⊃42·H+ complex (stabilized by the extra electrostatic attraction from the ionized moieties) manifests an increased emission with respect to free 42, as a result of the onset of AIE effects due to restricted rotation of the bound guest. Supramolecular nanoparticles (SNPs) were then prepared by nanoprecipitation of the complex, with the aim of making the aggregates more stable for potential biomedical applications. They were successfully tested for living-cell imaging, showing negligible cytotoxicity against both cancerous U87MG and normal HEK293 cells. In addition, when doxorubicin is encapsulated into SNPs, an energy transfer relay (ETR) effect (mediated by Förster resonance energy transfer and aggregation-derived quenching) turns off the fluorescence of both SNPs and doxorubicin.74In vitro studies showed that, after cellular internalization, protonation of doxorubicin in the acidic environment induces doxorubicin unloading, thus removing the ETR effect and restoring the AIE fluorescence of the nanoparticles and allowing the authors to reveal that the cargo molecules have been released into the nuclei.
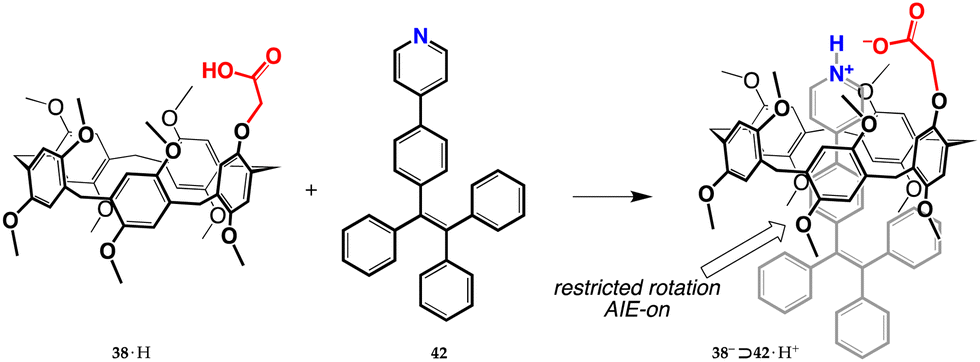 | ||
| Fig. 18 Formation of the AIE-active 38−⊃42·H+ complex.73 | ||
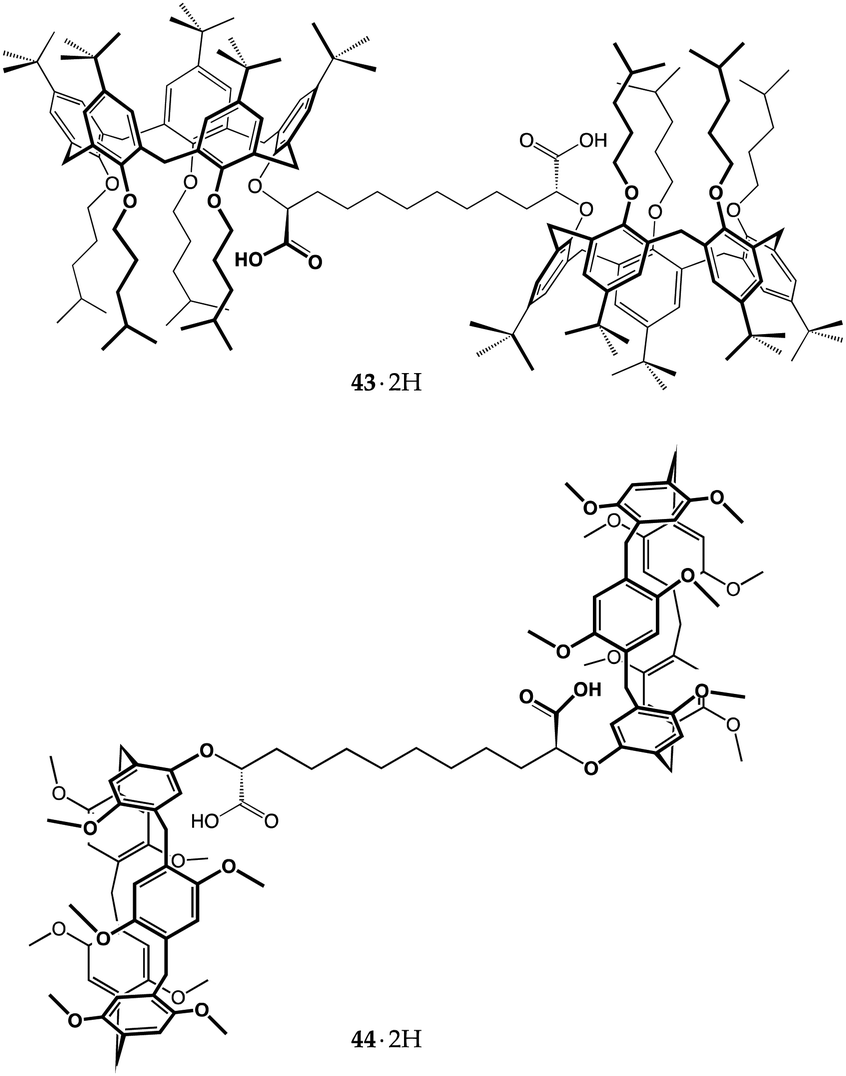
As a part of our ongoing studies on host–guest-based supramolecular polymers,75,76 we have recently taken advantage of the proton-transfer mediated recognition strategy to induce the self-assembly of large organized aggregates.77,78 The successful formation of these polymers relies on dynamic iterative recognition processes between the host and guest monomers. However, when the guest monomer includes a charged group, the counterion often plays a detrimental role on the efficiency of the self-assembly, ion-pairing with the guest moiety and depressing complexation.79,80 By choosing the right pair of ionizable complementary homoditopic monomers, we succeeded to harness internal ion pairing to produce overall neutral AA/BB-type supramolecular polymers composed of charged monomeric units. This was achieved by using dicarboxyl meso-bis-calix[5]arene 43·2H with divergent cavities and an α,ω-diamine.77
In the presence of equivalent amounts of 43·2H and 1,12-dodecanediamine, proton transfer triggered supramolecular polymerization. Diffusion Ordered SpectroscopY (DOSY) NMR studies, along with dynamic light scattering (DLS) investigations, demonstrated that, in the millimolar range, the [432−⊃+H3N(CH2)12NH3+]n polymer assembles into two families of cyclic aggregates, with average molecular masses of approximately 20 (ca. 8-mer) and 50 MDa (ca. 20000-mer), respectively. AFM micrographs showed the presence of torus-shaped aggregates with an average diameter of 100 nm (Fig. 19).77 The polymer disassembles upon the addition of a competitive chain stopper such as n-butylamine, providing bundles of fibres presumably composed of linear oligomers.
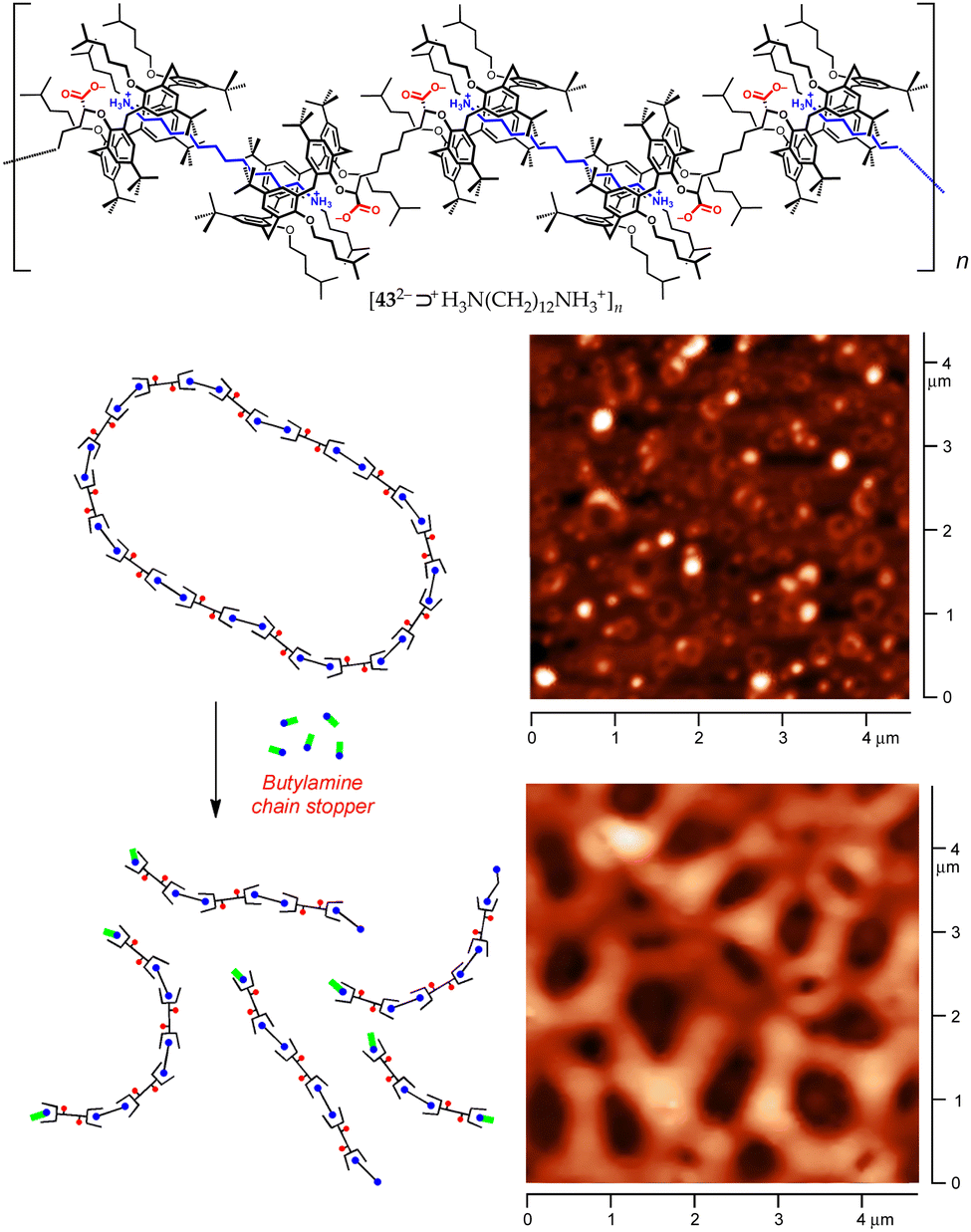 | ||
| Fig. 19 Top: Structure of the [432−⊃+H3N(CH2)12NH3+]n supramolecular polymer. Bottom: AFM micrographs demonstrating the disassembly of the polymer upon addition of n-BuNH2.77 | ||
The robustness of such a monomer design was tested on meso-bis-pillar[5]arene dicarboxylic acid 44·2H, by treating the latter with 1,12-dodecanediamine under identical conditions to those employed for bis-calixarene 43·2H. As expected, the host-to-guest proton transfer resulted in the self-assembly of the AA/BB-type supramolecular polymer [442−⊃+H3N(CH2)12NH3+]n. DOSY NMR analysis indicated that smaller aggregates were formed than those in the case of bis-calixarene 43·2H, averaging 46 kDa at 13 mM (Fig. 20).78 AFM analysis showed a picture more compatible with the presence of linear objects, forming a grid-like pattern with 50–150 nm thick strands, not much different than those seen for the disassembled 43·2H/H2N(CH2)12NH2/n-BuNH2 mixture (Fig. 20).
 | ||
| Fig. 20 Top: Structure of the [442−⊃+H3N(CH2)12NH3+]n supramolecular polymer. Bottom: AFM micrographs demonstrating the disassembly of the polymer upon addition of Et3N.78 | ||
Calix[5]arenes and pillar[5]arenes, however, profoundly differ in their mode of interaction with linear alkylammonium ions. The former are attracted to the charged ammonium terminus (O⋯HN+ contributing much more than CH⋯π interactions),52 whilst the latter relies predominantly on CH⋯π interactions with the alkyl chain.63 Not surprisingly, pillararenes are able to recognize linear (di)amines and alkylammonium and alkanediyldiammonium ions.66 In our case, the treatment of [442−⊃+H3N(CH2)12NH3+]n with Et3N caused – according to AFM (Fig. 20) and DOSY studies – the extensive disassembly of the original polymer, yielding low molecular-weight pillararene/dodecanediamine fragments held together by CH⋯π and weak COO−⋯H2N hydrogen-bond interactions.78 These data emphasize the critical contribution played by the electrostatic contribution to host–guest complexation provided by the groups ionized by proton transfer.
Future outlook
Recognition mediated by host-to-guest proton transfer and assisted by ion pairing is a phenomenon that has been present in supramolecular chemistry from the early days, having its roots planted in Cram's seminal work from almost half a century ago.15 It is surprising though that this conceptual approach has never been fully acknowledged as a regular tool for molecular recognition, and even more so given that Nature takes ample advantage of ion-pairing and salt-bridging interactions. This survey was carried out with the aim of organizing available data on this powerful yet often overlooked interaction and showing that the placement of a single acidic proton on a macrocyclic receptor scaffold may provide both additional strength and increased selectivity in the recognition of ammonium ions. We have also shown how this approach works across diverse molecular platforms – crown ethers, calixarenes, and pillararenes – and how it is already a core element in some current state-of-the-art applications: we therefore hope that proton transfer mediated complexation by ionizable receptors will be given a stable place among the design tools in amine recognition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge MIUR (Ministero dell’Istruzione, dell’Università e della Ricerca, PRIN_2009A5Y3N9 and PRIN_20179BJNA2 grants) and Università degli Studi di Messina for support.References
- B. Smith and L. Radom, J. Phys. Chem., 1991, 95, 10549–10551 CrossRef CAS.
- N. Eyet, S. Villano and V. Bierbaum, Int. J. Mass Spectrom., 2009, 283, 26–29 CrossRef CAS.
- E. Strittmatter, R. Wong and E. Williams, J. Phys. Chem. A, 2000, 104, 10271–10279 CrossRef CAS PubMed.
- S. Sen Gupta, J. Phys. Org. Chem., 2016, 29, 251–264 CrossRef CAS.
- S. Sen Gupta, J. Phys. Org. Chem., 2017, 30, e3605 CrossRef.
- G. Smith, Phys. Chem. Chem. Phys., 2018, 20, 18919–18923 RSC.
- S. Tripathi, S. Islam, S. Seth, A. Bauza, A. Frontera and S. Mukhopadhyay, CrystEngComm, 2020, 22, 8171–8182 RSC.
- S. Kumar and R. Nussinov, ChemBioChem, 2002, 3, 604–617 CrossRef CAS PubMed.
- J. Donald, D. Kulp and W. DeGrado, Proteins: Struct., Funct., Genet., 2011, 79, 898–915 CrossRef CAS PubMed.
- D. Anderson, W. Becktel and F. Dahlquist, Biochemistry, 1990, 29, 2403–2408 CrossRef CAS PubMed.
- S. Kumar and R. Nussinov, J. Mol. Biol., 1999, 293, 1241–1255 CrossRef CAS PubMed.
- H. Bosshard, D. Marti and I. Jelesarov, J. Mol. Recognit., 2004, 17, 1–16 CrossRef CAS PubMed.
- Z. Hendsch and B. Tidor, Protein Sci., 1994, 3, 211–226 CrossRef CAS PubMed.
- R. Bartsch, Solvent Extr. Ion Exch., 1989, 7, 829–854 CrossRef CAS.
- M. Newcomb and D. J. Cram, J. Am. Chem. Soc., 1975, 97, 1257–1259 CrossRef CAS.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 2495–2496 CrossRef CAS.
- S. S. Moore, T. L. Tarnowski, M. Newcomb and D. J. Cram, J. Am. Chem. Soc., 1977, 99, 6398–6405 CrossRef CAS.
- I. Goldberg, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2592–2600 CrossRef.
- M. A. McKervey and D. L. Mulholland, J. Am. Chem. Soc., Chem. Commun., 1977, 438–439 RSC.
- C. Browne, G. Ferguson, M. Mckervey, D. Mulholland, T. Oconnor and M. Parvez, J. Am. Chem. Soc., 1985, 107, 2703–2712 CrossRef CAS.
- T. W. Bell, P. G. Cheng, M. Newcomb and D. J. Cram, J. Am. Chem. Soc., 1982, 104, 5185–5188 CrossRef CAS.
- R. M. Izatt, J. S. Bradshaw, M. L. Colter, Y. Nakatsuji, N. O. Spencer, M. F. Brown, G. Arena, P. K. Tse and B. E. Wilson, J. Org. Chem., 1985, 50, 4865–4872 CrossRef CAS.
- J. S. Bradshaw, C. W. McDaniel, B. D. Skidmore, R. B. Nielsen, B. E. Wilson, N. K. Dalley and R. M. Izatt, J. Heterocycl. Chem., 1987, 24, 1085–1092 CrossRef CAS.
- J. Bradshaw, D. Chamberlin, P. Harrison, B. Wilson, G. Arena, N. Dalley, J. Lamb and R. Izatt, J. Org. Chem., 1985, 50, 3065–3069 CrossRef CAS.
- J. S. Bradshaw, G. E. Maas, J. D. Lamb, R. M. Izatt and J. J. Christensen, J. Am. Chem. Soc., 1980, 102, 467–474 CrossRef CAS.
- T. Kaneda, K. Sugihara, H. Kamiya and S. Misumi, Tetrahedron Lett., 1981, 22, 4407–4408 CrossRef CAS.
- K. Sugihara, T. Kaneda and S. Misumi, Heterocycles, 1982, 18, 57–61 CrossRef CAS.
- T. Kaneda, Y. Ishizaki, S. Misumi, Y. Kai, G. Hirao and N. Kasai, J. Am. Chem. Soc., 1988, 110, 2970–2972 CrossRef CAS.
- T. Kaneda, S. Umeda, Y. Ishizaki, H. Kuo, S. Misumi, Y. Kai, N. Kanehisa and N. Kasai, J. Am. Chem. Soc., 1989, 111, 1881–1883 CrossRef CAS.
- T. Kaneda, K. Hirose and S. Misumi, J. Am. Chem. Soc., 1989, 111, 742–743 CrossRef CAS.
- K. Fuji, K. Tsubaki, K. Tanaka, N. Hayashi, T. Otsubo and T. Kinoshita, J. Am. Chem. Soc., 1999, 121, 3807–3808 CrossRef CAS.
- K. Tsubaki, M. Nuruzzaman, T. Kusumoto, N. Hayashi, W. Bin-Gui and K. Fuji, Org. Lett., 2001, 3, 4071–4073 CrossRef CAS PubMed.
- K. Tsubaki, T. Kusumoto, N. Hayashi, M. Nuruzzaman and K. Fuji, Org. Lett., 2002, 4, 2313–2316 CrossRef CAS PubMed.
- J. H. Jung, S. J. Lee, J. S. Kim, W. S. Lee, Y. Sakata and T. Kaneda, Org. Lett., 2006, 8, 3009–3012 CrossRef CAS PubMed.
- G. Talanova, H. Hwang, V. Talanov and R. Bartsch, Chem. Commun., 1998, 419–420 RSC.
- R. A. Bartsch, H.-S. Hwang, V. S. Talanov, G. G. Talanova, D. W. Purkiss and R. D. Rogers, J. Org. Chem., 1999, 64, 5341–5349 CrossRef CAS.
- H. Zhou, K. Surowiec, D. W. Purkiss and R. A. Bartsch, Org. Biomol. Chem., 2005, 3, 1676–1684 RSC.
- J. P. Behr, J. M. Lehn, D. Moras and J. C. Thierry, J. Am. Chem. Soc., 1981, 103, 701–703 CrossRef CAS.
- S. Shinkai, Y. Shiramama, H. Satoh, O. Manabe, T. Arimura, K. Fujimoto and T. Matsuda, J. Chem. Soc., Perkin Trans. 2, 1989, 1167–1171 RSC.
- K. Ohto, M. Yano, K. Inoue, T. Yamamoto, M. Goto, F. Nakashio, S. Shinkai and T. Nagasaki, Anal. Sci., 1995, 11, 893–902 CrossRef CAS.
- T. Oshima, K. Inoue, K. Uezu and M. Goto, Anal. Chim. Acta, 2004, 509, 137–144 CrossRef CAS.
- S. Le Gac, M. Giorgi and I. Jabin, Supramol. Chem., 2007, 19, 185–197 CrossRef CAS.
- I. Batinic-Haberle, I. Spasojevic, Y. Jang, R. Bartsch and A. Crumbliss, Inorg. Chem., 1998, 37, 1438–1445 CrossRef CAS.
- T. Haino, T. Harano, K. Matsumura and Y. Fukazawa, Tetrahedron Lett., 1995, 36, 5793–5796 CrossRef CAS.
- T. Haino, K. Matsumura, T. Harano, K. Yamada, Y. Saijyo and Y. Fukazawa, Tetrahedron, 1998, 54, 12185–12196 CrossRef CAS.
- J. L. Atwood, L. J. Barbour and C. L. Raston, Cryst. Growth Des., 2002, 2, 3–6 CrossRef CAS.
- Y. Kubo, S. Maeda, S. Tokita and M. Kubo, Nature, 1996, 382, 522–524 CrossRef CAS.
- K. Tsubaki, T. Morimoto, T. Otsubo and K. Fuji, Org. Lett., 2002, 4, 2301–2304 CrossRef CAS PubMed.
- C. Reichardt, Angew. Chem., Int. Ed. Engl., 1979, 18, 98–110 CrossRef.
- F. Arnaud-Neu, S. Fuangswasdi, A. Notti, S. Pappalardo and M. Parisi, Angew. Chem., Int. Ed., 1998, 37, 112–114 CrossRef CAS.
- G. Gattuso, A. Notti, S. Pappalardo, M. Parisi, T. Pilati, G. Resnati and G. Terraneo, CrystEngComm, 2009, 11, 1204–1206 RSC.
- G. Gattuso, A. Notti, S. Pappalardo, M. F. Parisi, T. Pilati and G. Terraneo, CrystEngComm, 2012, 14, 2621–2625 RSC.
- C. Capici, G. Gattuso, A. Notti, M. Parisi, S. Pappalardo, G. Brancatelli and S. Geremia, J. Org. Chem., 2012, 77, 9668–9675 CrossRef CAS PubMed.
- G. Brancatelli, S. Pappalardo, G. Gattuso, A. Notti, I. Pisagatti, M. Parisi and S. Geremia, CrystEngComm, 2014, 16, 89–93 RSC.
- D. Garozzo, G. Gattuso, F. Kohnke, P. Malvagna, A. Notti, S. Occhipinti, S. Pappalardo, M. Parisi and I. Pisagatti, Tetrahedron Lett., 2002, 43, 7663–7667 CrossRef CAS.
- G. Brancatelli, G. Gattuso, S. Geremia, A. Notti, S. Pappalardo, M. Parisi and I. Pisagatti, Org. Lett., 2014, 16, 2354–2357 CrossRef CAS.
- G. Brancatelli, G. Gattuso, S. Geremia, N. Manganaro, A. Notti, S. Pappalardo, M. Parisi and I. Pisagatti, CrystEngComm, 2015, 17, 7915–7921 RSC.
- G. Brancatelli, G. Gattuso, S. Geremia, N. Manganaro, A. Notti, S. Pappalardo, M. Parisi and I. Pisagatti, CrystEngComm, 2016, 18, 5012–5016 RSC.
- S. Mecozzi and J. Rebek, Chem. – Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- T. Ogoshi, S. Kanai, S. Fujinami, T.-A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- P. Cragg and K. Sharma, Chem. Soc. Rev., 2012, 41, 597–607 RSC.
- M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed.
- T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS.
- G. Yu, B. Hua and C. Han, Org. Lett., 2014, 16, 2486–2489 CrossRef CAS PubMed.
- B. Hua and G. Yu, Tetrahedron Lett., 2014, 55, 6274–6276 CrossRef CAS.
- N. Strutt, R. Forgan, J. Spruell, Y. Botros and J. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668–5671 CrossRef CAS PubMed.
- B. Xia and M. Xue, Chem. Commun., 2014, 50, 1021–1023 RSC.
- C. Bruns, J. Stoddart and L. Fabbrizzi, Top. Curr. Chem., 2012, 323, 19–72 CrossRef CAS PubMed.
- H. Liang, B. Hua, F. Xu, L. Gan, L. Shao and F. Huang, J. Am. Chem. Soc., 2020, 142, 19772–19778 CrossRef CAS PubMed.
- J. Chen, J. Ding and T. Wei, Chem. Commun., 2021, 57, 9029–9039 RSC.
- Y. Zhou, H. Tang, Z. Li, L. Xu, L. Wang and D. Cao, Chem. Commun., 2021, 57, 13114–13117 RSC.
- P. Gilli, L. Pretto, V. Bertolasi and G. Gilli, Acc. Chem. Res., 2009, 42, 33–44 CrossRef CAS PubMed.
- D. Liu, J. Du, S. Qi, M. Li, J. Wang, M. Liu, X. Du, X. Wang, B. Ren, D. Wu and J. Shen, Mater. Chem. Front., 2021, 5, 1418–1427 RSC.
- X. Xue, S. Jin, C. Zhang, K. Yang, S. Huo, F. Chen, G. Zou and X. Liang, ACS Nano, 2015, 9, 2729–2739 CrossRef CAS PubMed.
- S. Pappalardo, V. Villari, S. Slovak, Y. Cohen, G. Gattuso, A. Notti, A. Pappalardo, I. Pisagatti and M. Parisi, Chem. – Eur. J., 2007, 13, 8164–8173 CrossRef CAS PubMed.
- G. Gattuso, A. Notti, A. Pappalardo, M. F. Parisi, I. Pisagatti, S. Pappalardo, D. Garozzo, A. Messina, Y. Cohen and S. Slovak, J. Org. Chem., 2008, 73, 7280–7289 CrossRef CAS PubMed.
- N. Manganaro, I. Pisagatti, A. Notti, A. Pappalardo, S. Patane, N. Micali, V. Villari, M. Parisi and G. Gattuso, Chem. – Eur. J., 2018, 24, 1097–1103 CrossRef CAS PubMed.
- A. Notti, I. Pisagatti, F. Nastasi, S. Patane, M. Parisi and G. Gattuso, J. Org. Chem., 2021, 86, 1676–1684 CrossRef CAS PubMed.
- C. Capici, Y. Cohen, A. D'Urso, G. Gattuso, A. Notti, A. Pappalardo, S. Pappalardo, M. Parisi, R. Purrello, S. Slovak and V. Villari, Angew. Chem., Int. Ed., 2011, 50, 11956–11961 CrossRef CAS.
- C. Gargiulli, G. Gattuso, A. Notti, S. Pappalardo and M. F. Parisi, Tetrahedron Lett., 2011, 52, 7116–7120 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |