
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolating elusive ‘Al(μ-O)M’ intermediates in CO2 reduction by bimetallic Al–M complexes (M = Zn, Mg)†
Matthew J.
Evans
a,
George H.
Iliffe
a,
Samuel E.
Neale
 b,
Claire L.
McMullin
*b,
J. Robin
Fulton
a,
Mathew D.
Anker
a and
Martyn P.
Coles
*a
b,
Claire L.
McMullin
*b,
J. Robin
Fulton
a,
Mathew D.
Anker
a and
Martyn P.
Coles
*a
aSchool of Chemical and Physical Sciences, Victoria University of Wellington, P.O. Box 600, Kelburn, Wellington, 6012, New Zealand. E-mail: martyn.coles@vuw.ac.nz
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: cm2025@bath.ac.uk
First published on 11th August 2022
Abstract
The reaction of compounds containing Al–Mg and Al–Zn bonds with N2O enabled isolation of the corresponding Al(μ-O)M complexes. Electronic structure analysis identified largely ionic Al–O and O–M bonds, featuring an anionic μ-oxo centre. Reaction with CO2 confirmed that these species correspond to the proposed intermediates in the formation of μ-carbonate compounds.
The synergistic combination of metal centres in a bimetallic complex can promote reactivity that is divergent from their monomeric counterparts.1 This effect is amplified in complexes containing direct metal–metal bonds,2 with early-late heterobimetallic compounds of the transition elements playing a central role in this field.3 As the main contributor to global warming, the transformation of carbon dioxide into value-added products has important societal benefits,4 and its two electron reduction via insertion into metal–metal bonds offers an attractive entry towards achieving this goal.5
Heterobimetallic complexes featuring polarised Al–M bonds have demonstrated the facile activation of CO2. This has been aided by the discovery of aluminyl anions,6 with systems supported by [4,5-(NDipp)2-2,7-tBu2-9,9-Me2-xanthene]2− (XanthNONDipp),7 and [(CH2SiMe2NDipp)2]2− (NC2NDipp)8 ligands dominant in this field (Fig. 1). The aluminium fragments have been partnered with Zn,9 and the coinage metals (Cu, Ag, Au)10 to afford complexes containing unsupported Al–M bonds (I). Reaction of I with CO2 proceeds with insertion into the Al–M bond to afford the corresponding dioxocarbene (II), the mechanism of which has been studied computationally.10c,d Species II can react with additional CO2 to afford the carbonate, IV, believed to proceed via an intermediate μ-oxo species III formed by the extrusion of CO. Examples of this putative intermediate have yet to be isolated in these systems, although other compounds containing the Al(μ-O)M motif have demonstrated this reactivity.11 We report herein the synthesis of Al–M (M = Zn, Mg) complexes derived from the [Al(NONDipp)]− aluminyl (NONDipp = [O(SiMe2NDipp)2]2−),12 and their reactivity with CO2, including isolation of the first μ-oxo intermediates (III). Although we acknowledge obvious differences in the chemistry of Mg and Zn compared with Cu, Ag and Au, we feel that the results presented here support the validity of the postulated mechanism of carbonate formation in the previously studied systems.
 | ||
| Fig. 1 Aluminyl derived Al–M systems that activate of CO2. Dipp = 2,6-iPr2C6H3. | ||
The reaction of [K{Al(NONDipp)}]2 with two equivalents of Mg(BDIMes)I(OEt2) or Zn(BDIMes)(μ-Cl)2Li(THF)2 afforded the bimetallic complexes (NONDipp)Al–M(BDIMes) (1-M, Scheme 1). The 1H NMR spectra showed the expected peaks for the (NONDipp)- and (BDIMes)-ligands in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, with broad resonances for the SiMe2 groups in 1-Zn suggesting restricted rotation about the Al–Zn bond. X-Ray diffraction studies show both systems crystallise as isostructural monomers containing unsupported Al–M bonds (Fig. 2). The Al–Zn (2.4860(5) Å) and Al–Mg (2.7711(6) Å) bonds are within the range of other reported Al–Mg (2.696(1)–2.7980(6) Å)7,8,13 and Al–Zn (2.448(2)–2.491(1) Å)9,13a–c,14 bonds.
:1 ratio, with broad resonances for the SiMe2 groups in 1-Zn suggesting restricted rotation about the Al–Zn bond. X-Ray diffraction studies show both systems crystallise as isostructural monomers containing unsupported Al–M bonds (Fig. 2). The Al–Zn (2.4860(5) Å) and Al–Mg (2.7711(6) Å) bonds are within the range of other reported Al–Mg (2.696(1)–2.7980(6) Å)7,8,13 and Al–Zn (2.448(2)–2.491(1) Å)9,13a–c,14 bonds.
 | ||
| Scheme 1 Synthesis of 1-Zn and 1-Mg. (i) Mg(BDIMes)I(OEt2) or [Li(THF)2][Zn(BDIMes)Cl2]. BDIMes = [HC{CMeNMes}2]−. | ||
 | ||
| Fig. 2 Thermal displacement plot (30% probability, H-atoms omitted, selected carbon atoms represented as spheres) of (a) 1-Zn (' = −x, y, ½ − z) and (b) 1-Mg. Selected bond lengths (Å): (a) Al–Zn 2.4860(5); (b) Al–Mg 2.7711(6). Bottom: Natural atomic charge data for the Al and M atoms (red = qAl/qM; blue = Δq = qAl − qM) {BP86/BS2; NBO7}. | ||
DFT calculations at the BP86/BS2 level of theory (see ESI†) and subsequent NBO (Natural Bonding Orbital) analysis of 1-Zn and 1-Mg highlights the electronic variance between these two species. The Wiberg Bond Index (WBI) values for the Al–M bonds suggest single bond character, which is stronger in 1-Zn (WBI(Al–Zn) = 0.740) than in 1-Mg (WBI(Al–Mg) = 0.602). Interestingly, natural atomic charge data suggest a reversal in the polarity of the Al–M bond based on the charge difference (Δq = qAl − qM, Fig. 2), with Al more electropositive than Zn in 1-Zn (Δq = +0.284), whereas Mg is the more electropositive partner in 1-Mg (Δq = −0.396). These data contrast those calculated for (XanthNONDipp)Al–M(BDIMes), which showed that Al was the less electropositive element in both Al–Mg (Δq = −0.69) and Al–Zn (Δq = −0.30) complexes.9
Further investigation of these charge differences revealed an appreciable basis set dependence on the qM values calculated for Al–M complexes. For example, we have performed calculations on the (XanthNONDipp) system using a triple-zeta basis set approach (6-311 + +G** = BS2), which show the same polarity direction for the Al–Mg bond as noted in 1-Mg. However, for the Al–Zn complex, the charge imbalance and bond polarity is reversed when compared with reported values calculated at the PBE0/SVP level (qAl = 0.96, qZn = 1.26 and Δq = −0.30), with our results showing values of qAl = 1.262, qZn = 1.001 giving a Δq = +0.261 at the BP86/BS2 level.9,15
Exposing a solution of 1-Zn to 1 bar of 13CO2 afforded (NONDipp)Al(μ-1κ2O,O′:2κC-O2C)Zn(BDIMes) (2, Scheme 2). A 13C{1H} resonance at δc 222.2 is characteristic of the carbenic carbon and reminiscent of other dioxocarbene complexes (range: δc 219.79 to δc 242.310c). The X-ray structure confirms the expected regiochemistry of the inserted CO2 and is consistent with other examples of II.9,10c,d The carbon–oxygen distances (range: 1.2917(18)–1.2970(18) Å) indicate delocalization, with relatively short Zn–C bonds.16
 | ||
| Scheme 2 Synthesis of 2 and 3. (i) 13CO2 (1 bar). Displacement ellipsoid plots of 3 (30% ellipsoids; H-atoms omitted; C-atoms represented as spheres). Selected bond lengths (Å) and angles (°): 2 {value from second molecule} C29–O2 1.2917(18) {1.292(2)}, C29–O3 1.2970(18) {1.2938(19)}, Al1–O2 1.8679(11) {1.8682(11)}, Al1–O3 1.8587(11) {1.8602(11)}, Zn1–C29 1.9641(14) {1.9640(15)}. 3 C1c–O1c 1.316(5), C1c–O2c 1.303(5), C1c–O3c 1.235(4), C2c–O4c 1.300(4), C2c–O5c 1.324(4), C2c–O6c 1.228(4). | ||
In contrast to 1-Zn, monitoring the reaction of 1-Mg with 13CO2 (1 bar) at room temperature by 13C NMR spectroscopy showed the immediate appearance of a peak at δC 184.5 corresponding to 13CO (Fig. S17, ESI†). This mirrors the reaction of (XanthNONDipp)Al–Cu(PtBu3) with CO2, which directly formed the carbonate complex IV at −78 °C (Fig. 1). The 13C{1H} NMR spectrum of crystals isolated from the reaction showed a 13C enriched peak at δC 164.1 consistent with [13CO3]2−. X-Ray diffraction of the product confirmed the formation of carbonate in {(NONDipp)Al(μ-1κ2O,O′:2κO′′-CO3)}2{Mg(THF)4} (3, Scheme 2).
Each aluminium is O,O′-coordinated to a planar [CO3]2− ligand. The remaining oxygen atom of each carbonate is trans-bound to an octahedral magnesium, with four molecules of THF completing the coordination sphere. A plausible rationale for this result is that the reaction proceeds via the monocarbonate (NONDipp)Al(CO3)Mg(BDIMes), which exists in a Schlenk-type equilibrium with 3 and Mg(BDIMes)2,17 although we were unable to isolate the homoleptic magnesium product in this study.
Previous work with potassium aluminyl systems identified that formation of carbonate compounds from CO2 proceeded via monoalumoxane anions containing terminal Al–O bonds.18 These species were isolated as [Al(XanthNONDipp)(O)]− and [Al(NONDipp)(O)]− from the reaction of the corresponding potassium aluminyl with N2O gas. Analogous compounds have been inferred as intermediates in the conversion of CO2 to carbonate in bimetallic Al–M (M = Cu, Ag, Au) systems (III, Fig. 1), although no examples have been isolated.19 We therefore targeted the isolation of such compounds containing the Al(μ-O)M group to allow us to gain a better understanding of the bonding and to unequivocally demonstrate the viability of this pathway in our systems.
The reaction of 1-Zn and 1-Mg with N2O (1 bar) proceeded to afford (NONDipp)Al(μ-O)M(BDIMes) (4-Zn and 4-Mg, Scheme 3). The 1H NMR resonances of the SiMe2 groups are no longer broad (as noted for 1-Zn and 1-Mg), consistent with an increased distance between the NONDipp- and BDIMes-ligands and greater rotational freedom. X-Ray crystallography confirmed formation of the bimetallic complexes containing a μ-oxo ligand (Fig. 3). Each metal is three-coordinate, with an unsupported, approximately linear bridging oxo-ligand (Al–O–M range: 173.58(11)°–176.94(12)°). The Al–O bond lengths (range: 1.6413(16)–1.6472(15) Å) are considerably shorter than the sum of the molecular covalent double bond radii, r2 (Σ(r2)Al,O = 1.70 Å) and almost indistinguishable from that noted in the terminal monoalumoxane anion, [Al(NONDipp)(O)]− (1.6362(14) Å).18b Significant differences are apparent when comparing the bonding in 4-Zn and 4-Mg with other Al(μ-O)M groups (M = Zn,20M = Mg21) involving tetrahedral Al. In previous examples, the Al–O bond is longer (range: 1.671(2)–1.682(2) Å) with a significant bending at the μ-oxide atom (Al–O–M range: 144.78(15)°–159.6(1)°). These variations prompted a computational investigation of the electronic structure of the Al(μ-O)M group in 4-Zn and 4-Mg.
 | ||
| Scheme 3 Synthesis of 4-Zn, 4-Mg and 5. (i) N2O (1 bar); (ii) 13CO2 (1 bar). | ||
 | ||
| Fig. 3 Thermal displacement plot (30% probability, H-atoms omitted, selected carbon atoms represented as spheres) of (a) 4-Zn and (b) 4-Mg. Selected bond lengths (Å) and angles (°) {value from second molecule}: (a) Al1–O2 1.6413(16) {1.6428(15)}, Zn1–O2 1.7826(15) {1.7867(15)}; Al1–O2–Zn1 176.94(12) {173.74(11)}. (b) Al1–O2 1.6472(15) {1.6426(17)}, Mg1–O2 1.8004(16) {1.8009(18)}; Al1–O2–Mg1 173.58(11) {174.41(14)}. | ||
QTAIM and NBO analyses were performed at the BP86/BS2 level on 4-Zn and 4-Mg to interrogate the Al–O and O–M bonds. For both complexes, NBO7 charges show that the Al centre was more electropositive than M, although this charge difference was more notable for 4-Zn (Δq = +0.596) than 4-Mg (Δq = +0.400). QTAIM analysis identifies appreciable and similar Al–O bond critical points (BCPs) for each complex (ρ(r): 4-Zn = 0.101 eÅ−3; 4-Mg = 0.104 eÅ−3), where positive Laplacian values (∇2ρ(r): 4-Zn = 0.858 eÅ−5; 4-Mg = 0.875 eÅ−5) and weakly positive energy densities, H(r), and |V(r)| < 2G(r),22 indicate an Al–O bond with ionic character in both complexes (Table S5, ESI†). This is further supported by low WBI(Al–O) values (4-Zn = 0.372; 4-Mg = 0.426) when compared with the [Al(NONDipp)(O)]− anion (WBI(Al–O) = 1.11).23 Moreover, we note that the values of ρ(r) are similar to that of the monomeric [Al(NONDipp)(O)]− anion (ρ(r) = 0.115 eÅ−3; ∇2ρ(r) = 0.990 eÅ−5),23 indicating that the terminal M(BDIMes) group only minimally perturbs the charge density and electronic composition of the Al–O bond. A more pronounced difference is observed for the O–M BCPs in each compound. For 4-Zn, the O–Zn bond has more electron density at the BCP (ρ(r) = 0.116 eÅ−3) than the adjacent Al–O BCP with a delocalization index DI(Zn|O) = +0.710 compared with DI(Al|O) = +0.404, suggesting the interaction is less ionic than Al–O. For 4-Mg, however, a much lower density is captured at the O–Mg BCP (ρ(r) = 0.066 eÅ−3), with a DI(Mg|O) = +0.270.
NBO analysis of 4-Zn and 4-Mg support the QTAIM analyses. Firstly, the NBO search in each complex generated an optimal Lewis structure essentially consisting of a series of 2s and 2pn lone pairs on a μ-oxo ion. Subsequent second-order perturbation energy analysis of the Fock matrix in the NBO basis reveals that the Al–O interaction consists of a suite of appreciable O → Al donor–acceptor NBO interactions, in which the 2s and 2pn donor NBOs on the bridging μ-oxo ion (LP2px, LP2py, LP2pz) interact with vacant 3s and 3pn acceptor NBOs at the Al centre. Moreover, the O–M interaction is identified to be much stronger in 4-Zn than 4-Mg, with a significant O → Zn donor–acceptor interaction of 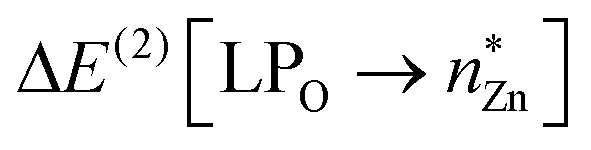 = 49.3 kcal mol−1 (Fig. 4).
= 49.3 kcal mol−1 (Fig. 4).
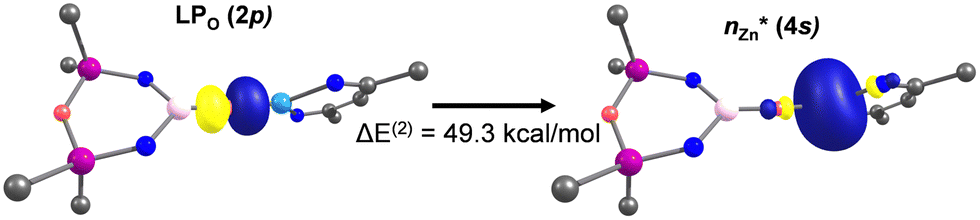 | ||
| Fig. 4 The strongest donor–acceptor interaction between the bridging oxo and Zn identified via NBO in 4-Zn. | ||
Conversion of the dioxocarbene (XanthNONDipp)Al(μ-O2C)M(PtBu3) to the carbonate species occurs under an atmosphere of CO2 at −78 °C (M = Cu) or 80 °C (M = Ag), whereas the Al/Au analogue is stable.10c The (NC2NDipp) analogues were less reactive, with the corresponding Al(μ-O2C)Cu requiring heating to 60 °C.10d To investigate this rection with our systems, a solution of 2 was heated to 353 K under an atmosphere of 13CO2. However, no further reaction was noted, potentially reflecting the high carbophilicity of zinc. In contrast, a solution of the μ-oxide complex 4-Zn reacted instantly with 13CO2 to afford the carbonate product, (NONDipp)Al(μ-CO3)Zn(BDIMes) (5). 13C{1H} NMR spectroscopy supports the formation of the carbonate product (δC 168.4) and a X-ray diffraction study confirms a 1κ2O,O′:2κO′′-bridging mode (Fig. S29, ESI†). These observations suggest the loss of CO from 2 to form 4-Zn is energetically disfavoured, although addition to the μ-oxide to form the carbonate is a low energy process. The corresponding reaction of the magnesium complex 4-Mg with CO2 gave the carbonate product 3.
In summary, we have shown that isolated Al(μ-O)M species react with CO2 to form the carbonate product, confirming their role as intermediates in this reaction sequence.
M. P. C. and M. J. E. acknowledge Government funding from the Marsden Fund Council from, managed by Royal Society Te Apārangi (Grant Number: MFP-VUW2020). C. L. M. and S. E. N. acknowledge funding from the EPSRC (EP/R020752). This research made use of the Anatra, Balena (Bath) and Rāpoi (VUW) High Performance Computing (HPC) Services. Data used within this publication can be accessed from the Research Data Service – https://library.bath.ac.uk/research-data.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. M. Gil-Negrete and E. Hevia, Chem. Sci., 2021, 12, 1982–1992 RSC; (b) T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021, 60, 9247–9262 CrossRef CAS PubMed; (c) J. Campos, Nat. Rev. Chem., 2020, 4, 696–702 CrossRef CAS; (d) S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS PubMed.
- I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef CAS.
- (a) E. Bodio, M. Picquet and L. Gendre, in Topics in Organometallic Chemistry, ed., P. Kalck, Springer International Publishing, Switzerland, 2016, vol. 59, pp. 139–186 Search PubMed; (b) N. Wheatley and P. Kalck, Chem. Rev., 1999, 99, 3379–3420 CrossRef CAS PubMed.
- X. Wang, C. Xia and L. Wu, Green Chem., 2018, 20, 5415–5426 RSC.
- (a) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS; (b) X. Yin and J. R. Moss, Coord. Chem. Rev., 1999, 181, 27–59 CrossRef CAS; (c) W. Leitner, Coord. Chem. Rev., 1996, 153, 257–284 CrossRef CAS.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS.
- R. J. Schwamm, M. P. Coles, M. S. Hill, M. F. Mahon, C. L. McMullin, N. A. Rajabi and A. S. S. Wilson, Angew. Chem., Int. Ed., 2020, 59, 3928–3932 CrossRef CAS PubMed.
- M. M. D. Roy, J. Hicks, P. Vasko, A. Heilmann, A.-M. Baston, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 22301–22306 CrossRef CAS PubMed.
- (a) J. Hicks, A. Mansikkamäki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237–241 CrossRef CAS PubMed; (b) H.-Y. Liu, R. J. Schwamm, M. S. Hill, M. F. Mahon, C. L. McMullin and N. A. Rajabi, Angew. Chem., Int. Ed., 2021, 60, 14390–14393 CrossRef CAS PubMed; (c) C. McManus, J. Hicks, X. Cui, L. Zhao, G. Frenking, J. M. Goicoechea and S. Aldridge, Chem. Sci., 2021, 12, 13458–13468 RSC; (d) H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon and C. L. McMullin, Dalton Trans., 2022, 51, 3913–3924 RSC.
- (a) C. Weetman, A. Porzelt, P. Bag, F. Hanusch and S. Inoue, Chem. Sci., 2020, 11, 4817–4827 RSC; (b) C. Weetman, P. Bag, T. Szilvási, C. Jandl and S. Inoue, Angew. Chem., Int. Ed., 2019, 58, 10961–10965 CrossRef CAS PubMed; (c) J. A. Castro-Osma, M. North, W. K. Offermans, W. Leitner and T. E. Müller, ChemSusChem, 2016, 9, 791–794 CrossRef CAS PubMed.
- R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed.
- (a) C. Bakewell, B. J. Ward, A. J. P. White and M. R. Crimmin, Chem. Sci., 2018, 9, 2348–2356 RSC; (b) S. Brand, H. Elsen, J. Langer, S. Grams and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 15496–15503 CrossRef CAS PubMed; (c) A. Paparo, C. D. Smith and C. Jones, Angew. Chem., Int. Ed., 2019, 58, 11459–11463 CrossRef CAS PubMed; (d) A. Friedrich, J. Eyselein, J. Langer, C. Färber and S. Harder, Angew. Chem., Int. Ed., 2021, 60, 16492–16499 CrossRef CAS PubMed.
- J. Weßing, C. Göbel, B. Weber, C. Gemel and R. A. Fischer, Inorg. Chem., 2017, 56, 3517–3525 CrossRef PubMed.
- To examine the factors that determine the charges calculated for Al–M systems, a more detailed study is underway investigating the role of different levels of theory and QC charge schemes on the computed values.
- (a) P. Jochmann and D. W. Stephan, Chem. – Eur. J., 2014, 20, 8370–8378 CrossRef CAS PubMed; (b) M. E. Grundy, K. Yuan, G. S. Nichol and M. J. Ingleson, Chem. Sci., 2021, 12, 8190–8198 RSC.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem. – Eur. J., 2010, 16, 938–955 CrossRef CAS PubMed.
- (a) J. Hicks, A. Heilmann, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 17265–17268 CrossRef CAS PubMed; (b) M. D. Anker and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 18261–18265 CrossRef CAS PubMed.
- The reaction of (xanthNONDipp)Al–Ag(PtBu3) with N2O generated the μ-O bridged species in situ. However the complex proved “too thermally fragile to be isolated”. See reference 10(c).
- S. Schulz, J. Spielmann, D. Bläser and C. Wölper, Chem. Commun., 2011, 47, 2676–2678 RSC.
- (a) S. Nembenna, H. W. Roesky, S. K. Mandal, R. B. Ostwald, A. Pal, R. Herbst-Irmer, M. Noltemeyer and H.-G. Schmidt, J. Am. Chem. Soc., 2006, 128, 13056–13057 CrossRef CAS PubMed; (b) S. Nembenna, S. Singh, S. S. Sen, H. W. Roesky, H. Ott and D. Stalke, Z. Anorg. Allg. Chem., 2011, 637, 201–205 CAS.
- P. S. V. Kumar, V. Raghavendra and V. Subramanian, J. Chem. Sci., 2016, 128, 1527–1536 CrossRef CAS.
- M. J. Evans, M. D. Anker, C. L. McMullin, S. E. Neale, N. A. Rajabi and M. P. Coles, Chem. Sci., 2022, 13, 4635–4646 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data; full details of computational experiments. CCDC 2183690–2183696. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04028j |
| This journal is © The Royal Society of Chemistry 2022 |