
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New route to amide-functionalized N-donor ligands enables improved selective solvent extraction of trivalent actinides†
Rachel
Bulmer
a,
Thomas B.
Spencer
a,
Andreas
Wilden
 b,
Giuseppe
Modolo
b,
Trong-Hung
Vu
c,
Jean-Pierre
Simonin
c and
Frank W.
Lewis
*a
b,
Giuseppe
Modolo
b,
Trong-Hung
Vu
c,
Jean-Pierre
Simonin
c and
Frank W.
Lewis
*a
aDepartment of Applied Sciences, Faculty of Health and Life Sciences, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK. E-mail: frank.lewis@northumbria.ac.uk
bForschungszentrum Jülich GmbH, Institut für Energie und Klimaforschung – Nukleare Entsorgung und Reaktorsicherheit (IEK-6), 52428, Jülich, Germany
cLaboratoire PHENIX, CNRS, Sorbonne Université (Campus P.M. Curie), 4 Place Jussieu, Case 51, F-75005, Paris, France
First published on 31st August 2022
Abstract
A new general synthetic route to selective actinide extracting ligands for spent nuclear fuel reprocessing has been established. The amide-functionalized ligands separate Am(III) and Cm(III) from the lanthanides with high selectivities and show rapid rates of metal extraction. The ligands retain the advantages of the analogous unfunctionalized ligands derived from camphorquinone, whilst also negating their main drawback; precipitate formation when in contact with nitric acid. These studies could enable the design of improved solvent extraction processes for closing the nuclear fuel cycle.
Some of the main contributors to the long-term radiotoxicity of spent fuel arising from nuclear electricity production are the minor actinides americium, curium and neptunium. After reprocessing to remove uranium and plutonium in the PUREX process,1 the remaining spent fuel remains radiotoxic for ca. 104 years.2 If the minor actinides are also removed, the remaining material would take only a few hundred years to decay to the levels of natural uranium and its heat load would decrease significantly. As nuclear energy expands worldwide,3 it becomes imperative to develop viable options for future reprocessing to remove these elements prior to geological disposal of the remaining waste.4
To accomplish this, a solvent extraction process is required that can extract the minor actinides from nitric acid solution and separate them from the chemically similar, less-radiotoxic lanthanide fission products prior to their burn-up in advanced reactors or accelerator-driven systems.5 Many soft N- and S-donor ligands that can discriminate between the more extended 5f orbitals of the actinides and the more contracted 4f orbitals of the lanthanides have been evaluated for this separation.6,7 Bis-1,2,4-triazine ligands such as 1–3 (Fig. 1) fulfil many of the challenging criteria for use in such a separation process, and are among the current N-donor ligands of choice for further development.8
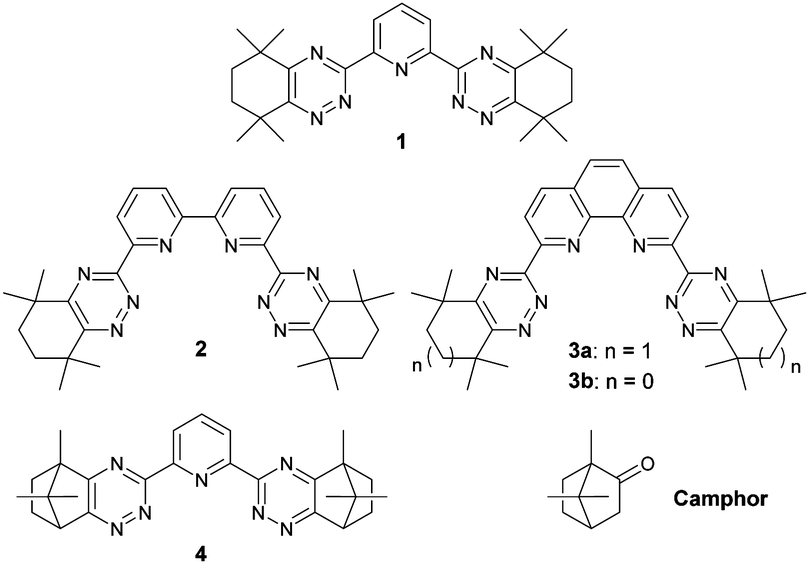 | ||
| Fig. 1 Structures of bis-1,2,4-triazine N-donor ligands 1–4 and camphor. | ||
More recently, ligand 4 derived from camphor (Fig. 1) was disclosed as a highly promising actinide selective extracting agent.9 In particular, 4 showed significantly higher solubilities than ligands 1–3 in diluents compatible with nuclear reprocessing, and the rates of metal extraction for ligand 4 were significantly faster than for ligands 2 and 3. However, in contrast to ligands 1–3, solutions of 4 formed precipitates in contact with nitric acid solutions of high concentrations, which renders 4 unsuitable for further process development. The precipitate formation observed is thought to be due to competing protonation of 4 and precipitation of the protonated ligand.
We proposed that more lipophilic derivatives of 4 and related ligands would be less likely to form precipitates in contact with nitric acid, whilst also retaining the advantages of 4. Since derivatives of camphor (Fig. 1) can be readily functionalized at each of its three methyl groups,10 we decided to explore functionalized derivatives of camphor as synthetic precursors to new, more lipophilic bis-1,2,4-triazine ligands. In this communication, we present our preliminary results on novel, amide-functionalized ligands derived from a functionalized camphor derivative.
We chose enantiomerically pure and inexpensive (+)-10-camphorsulfonyl chloride 5 as the starting material for the synthesis of the novel diketones to avoid the possibility of diastereomer formation during ligand synthesis, and to maximize the extraction of metal ions.11 Compound 6 was synthesized in 73% yield from 5 following the literature procedure,12 and converted into the known diketone 7 in 98% yield.13 The novel diketones 8–10 were then obtained from 7 and three representative secondary amines in 86–96% yields (Scheme 1).
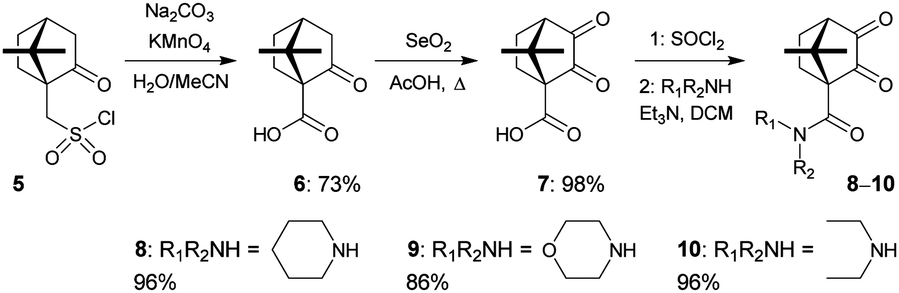 | ||
| Scheme 1 Synthesis of diketones 8–10 from (+)-10-camphorsulfonyl chloride 5. | ||
We next explored the synthesis of novel ligands by condensation reactions of diketones 8–10 with the known bis-amidrazone 11.8c Condensation reactions of 11 with diketones 8–10 in acetic acid cleanly gave the novel ligands 12–14 in moderate yields (Scheme 2). Use of ethanol or 1,4-dioxane as solvents resulted in incomplete conversion to the products.
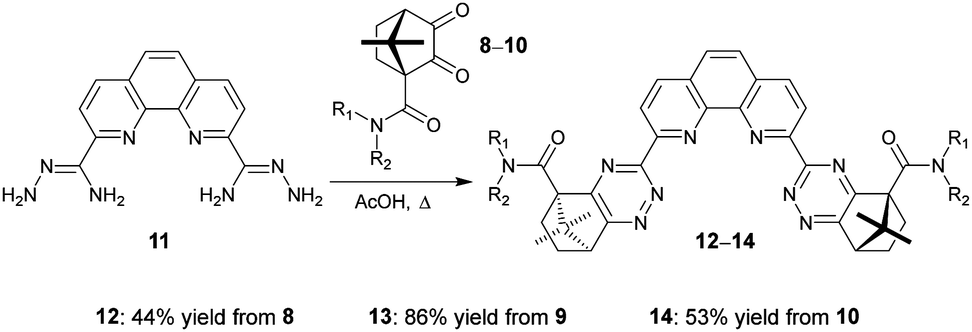 | ||
| Scheme 2 Synthesis of ligands 12–14 from diketones 8–10. | ||
We also decided to synthesize ligands 17 and 18 in order to determine what effect, if any, the addition of an amide functionality onto the aliphatic part would have on the minor actinide extraction properties of these ligands. The synthesis of 18 was previously reported14 but its minor actinide extraction properties have not been determined. Thus, condensation reaction of enantiomerically pure (+)-camphorquinone 16 with each of 15 and 11 in acetic acid afforded the novel ligands 17 and 18 in 53% and 59% yields, respectively (ESI†).
The measured solubilities of ligands 12–14 in 1-octanol ranged from 40.9–50.4 mM (ESI†). These are significantly higher than the maximum solubilities of 2 and 3a in the same diluent (10 mM15 and 15 mM,8c respectively), but significantly lower than the solubility of 4 (200 mM).9 The measured solubility of 17 in 1-octanol was 58.1 mM.
With ligands 12–14, 17 and 18 in hand, we next studied their ability to extract and separate Am(III) and Cm(III) from Eu(III) and the lanthanides in the SANEX process. Nitric acid solutions spiked with 241Am(III), 244Cm(III) and 152Eu(III) radionuclides containing all the lanthanides (except Pm) were contacted with 10 mM solutions of the ligands 12–14, 17 and 18 in 1-octanol for 1 hour, and distribution ratios (D) for metal ions were measured by α- and γ-spectroscopy or ICP-MS. For ligand 12, an effective separation of Am(III) and Cm(III) from Eu(III) and other lanthanides was observed (Fig. 2). The maximum D values for Am(III) and Cm(III) were 55 and 17, respectively, at ≥1 M HNO3, while the D values for Eu(III) were less than 1 across the range of nitric acid concentrations. The separation factor for Am(III) over Eu(III) (SFAm/Eu) increased with increasing nitric acid concentration to a maximum value of 231. The maximum distribution ratios for Am(III) and Cm(III) for 12 were significantly lower than those of 3a (DAm ≤ 1000)8c and about half as high as those of 3b (DAm ≤ 100).16 The extraction results for ligand 14 were broadly comparable to those of 12, although this ligand was somewhat less selective than 12 (SFAm/Eu ≤ 112, ESI†).
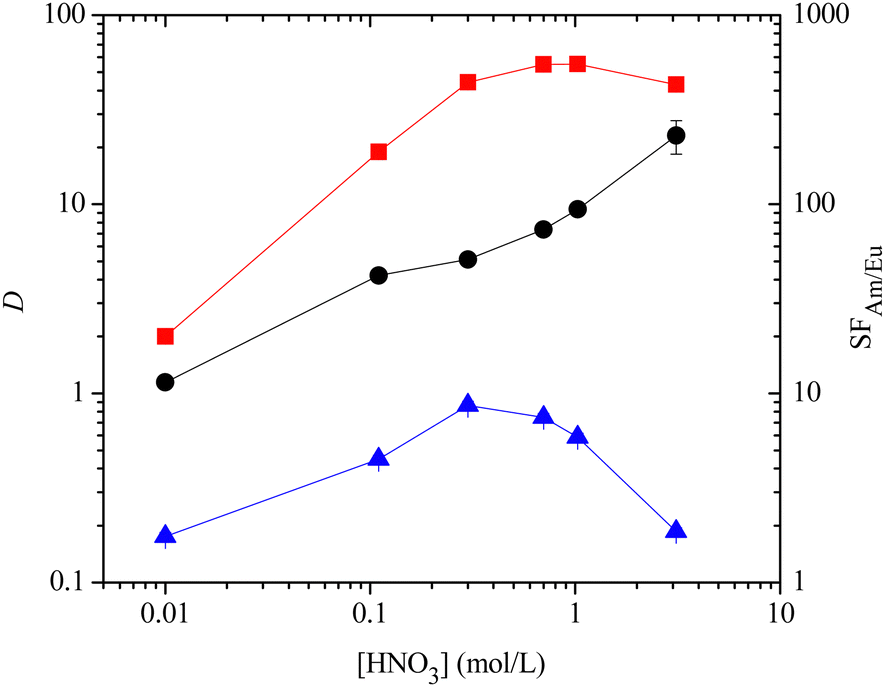 | ||
Fig. 2 Extraction of Am(III) and Eu(III) by ligand 12 (0.01 M) into 1-octanol as a function of the initial nitric acid concentration of the aqueous phase (D = distribution ratio, SF = separation factor,  = DAm, = DAm,  = DEu, ● = SFAm/Eu, contact time: 60 min, temperature: 22 °C ± 1 °C). = DEu, ● = SFAm/Eu, contact time: 60 min, temperature: 22 °C ± 1 °C). | ||
Interestingly, no precipitate formation was observed for ligands 12 and 14 in any of the extraction experiments, in contrast to 4.9 However, extensive precipitate formation was observed for 13 in the extraction experiments at all nitric acid concentrations, and this ligand did not extract Am(III) or Cm(III) from nitric acid into 1-octanol (DAm < 1, ESI†).
We next probed the rates of extraction of Am(III), Cm(III) and Eu(III) by ligands 12–14 into 1-octanol. Rapid rates of metal extraction are desirable in a solvent extraction process so that equilibrium can be reached within short contact times. The D values for the extraction of Am(III), Cm(III) and Eu(III) from 1.0 M HNO3 by ligands 12 and 14 as a function of contact time are presented in the ESI.† Both ligands 12 and 14 showed rapid rates of metal extraction and equilibrium D values were already reached for all metal ions within 5 minutes of phase mixing in the absence of a phase-modifier. These rates of metal extraction are significantly faster than those of ligands 215 and 3a8c (which require 60 minutes and 15 minutes of phase mixing, respectively, to reach equilibrium), and are comparable to those of ligand 4.9
We also measured the D values for Am(III), Cm(III) and Eu(III) for the parent ligands 17 and 18 derived from (+)-camphorquinone 16 (ESI†). For 17, an effective separation of Am(III) and Cm(III) from Eu(III) and other lanthanides was observed at nitric acid concentrations between 0.8–1.0 M HNO3 (DAm ≥ 3, DEu ≤ 0.06, SFAm/Eu = 133–153). The D values for 18 were significantly higher than those of 17, and 18 showed an effective and highly selective separation of Am(III) and Cm(III) from Eu(III) and other lanthanides across the range of nitric acid concentrations (DAm ≥ 46, DEu ≤ 0.23, SFAm/Eu ≤ 265). For both ligands 17 and 18 however, there was a significant drop in the D values for all metal ions at 3 M HNO3 and there was significant precipitate formation in the extraction experiments at all nitric acid concentrations. This was presumably due to precipitation of the protonated ligand, as observed previously with ligand 4. Thus, it appears all three parent ligands 4, 17 and 18 derived from (+)-camphorquinone 16 are susceptible to precipitate formation in contact with nitric acid, in contrast to the amide-functionalized ligands 12 and 14.
We then carried out NMR titrations with La(III), Lu(III) and Y(III) (as nitrate salts) to probe for differences in metal speciation between the camphor-derived ligands 12, 14 and 18, and the analogous ligands 3a and 3b reported previously.16,17 A single complex species was observed initially during the titrations of 12 and 14 with each metal, and the complete disappearance of the free ligand resonances at a metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand ratio of ca. 0.5 in each case indicates this was the expected 1:2 complexes [M(L)2(NO3)]2+ (L = ligand), in agreement with previous results for ligands 3a17 and 3b.16 However, in contrast to these ligands, the 1:1 complexes of 12 and 14 predominated at higher metal:ligand ratios and, in the case of La(III), the 1:2 complexes of 12 and 14 disappeared completely at the end of the titrations. One exception was the titration of 14 with Y(III), where 50% of the 1:2 complex was present at a metal:ligand ratio of 1.2. However, when the titration was resumed after one week, all of the 1:2 complex had completely dissociated. We attribute this to the relative kinetic inertness of Y(III) toward ligand substitution18 compared to La(III) and Lu(III), which was observed previously in NMR titrations of 3a and 3b.16,17
:ligand ratio of ca. 0.5 in each case indicates this was the expected 1:2 complexes [M(L)2(NO3)]2+ (L = ligand), in agreement with previous results for ligands 3a17 and 3b.16 However, in contrast to these ligands, the 1:1 complexes of 12 and 14 predominated at higher metal:ligand ratios and, in the case of La(III), the 1:2 complexes of 12 and 14 disappeared completely at the end of the titrations. One exception was the titration of 14 with Y(III), where 50% of the 1:2 complex was present at a metal:ligand ratio of 1.2. However, when the titration was resumed after one week, all of the 1:2 complex had completely dissociated. We attribute this to the relative kinetic inertness of Y(III) toward ligand substitution18 compared to La(III) and Lu(III), which was observed previously in NMR titrations of 3a and 3b.16,17
Interestingly, examination of the aromatic region of the 1H NMR spectra at the end of the titrations of 12 and 14 with each metal revealed the presence of three additional complex species, instead of the one additional species expected for the neutral 1:1 complex [M(L)(NO3)3] previously observed with ligands 3a and 3b.16,17 Since only 1:1 metal:ligand complexes are formed at higher metal:ligand ratios by dissociation of the 1:2 complexes,16,17 these species were assigned to the three 1:1 complexes of 12 and 14 that are theoretically possible; the symmetrical 1:1 complex involving tetradentate coordination of the ligand (species A; formed by initial dissociation of the 1:2 complex), the unsymmetrical 1:1 complex involving pentadentate coordination of the ligand (species B) and the symmetrical 1:1 complex involving hexadentate coordination of the ligand (species C). The speciation of ligand 14 with La(III) is shown in Fig. 3, and the structures of the different 1:1 complex species are shown in the ESI.†
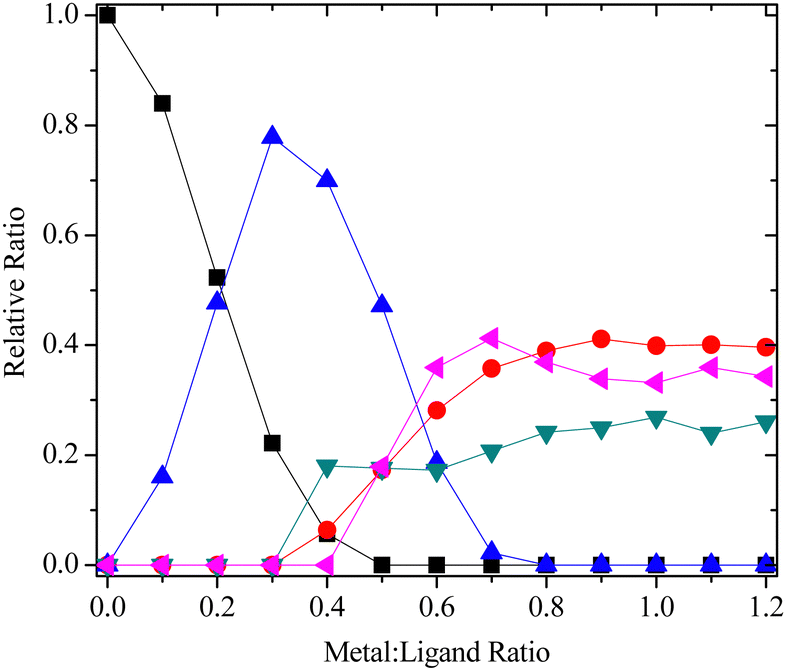 | ||
Fig. 3 Species distribution for the 1H NMR titration of 14 with La(NO3)3 in CD3CN (■ = free ligand,  = 1:2 complex, = 1:2 complex,  = species A, = species A,  = species B, = species B,  = species C). = species C). | ||
Species B and C are formed when the N4 nitrogen of the triazine ring and amide carbonyl O atom both coordinate to the metal instead of the N2 nitrogen of the triazine ring. Complexation via N4 is highly unusual in metal complexes of bis-1,2,4-triazine ligands, and previous structural evidence shows it is always the N2 nitrogen of the triazine ring that coordinates to the metal.19 To our knowledge, there is only one isolated report in the literature describing coordination of bis-1,2,4-triazine ligands with lanthanides via the N4 atom.20 Although species B and C are presumably less stable than species A, this is offset by the increase in ligand denticity that results when the amide carbonyl O atom also coordinates to the metal.
In the 1H NMR titrations of ligand 18 with La(III), Lu(III) and Y(III), only the expected 1:2 and 1:1 complexes were observed initially, in agreement with previous results.16,17 However, close examination of the 1H NMR spectra showed minor amounts (≤10%) of a third complex species, which was tentatively assigned as the unsymmetrical 1:1 complex where the N4 atom of one of the two triazine rings coordinates to the metal. This 1:1 complex species was not as prominent as the analogous 1:1 complex species B and C observed with ligands 12 and 14, presumably due to the absence of the additional ligating O atom in 18 that is present in 12 and 14.
On comparing the species distribution of metal complexes of ligands 12, 14 and 18 with those of the related ligands 3a and 3b with the same metals (ESI†), it is evident that the hydrophobic 1:2 complexes [M(L)2(NO3)]2+ are significantly less favoured for the camphor-derived ligands 12, 14 and 18 than for ligands 3a and 3b. Since it is these 1:2 complexes that are extracted into the organic phase, this could explain why the D values observed for Am(III), Cm(III) and Eu(III) in the extraction experiments were consistently lower with the camphor-derived ligands 12, 14 and 18 than with 3a and 3b.8c,16
To uncover the reasons for the rapid rates of metal extraction observed for ligands 12 and 14, we measured the interfacial tensions between aqueous 1 M nitric acid solutions and solutions of 12 in 1-octanol using the du Noüy ring method, and compared them to the previous results for 3a.8c The decrease in interfacial tension as the ligand concentration increases clearly shows that ligand 12 is surface active at the interface, in agreement with the rapid rates of metal extraction observed and previous measurements with 3a (ESI†). We then compared the extraction kinetics of Eu(III) and Am(III) of ligands 12 and 3a dissolved in 1-octanol using the rotating membrane cell method (ESI†).8c The Eu(III) extraction and back-extraction rate constants for 12 are significantly larger than the Eu(III) extraction rate constant for 3a, both in the absence and presence of N,N,N′,N′-tetraoctyldiglycolamide (TODGA) as phase modifier (ESI†). In addition, the Am(III) extraction rate constant for ligand 12 is significantly larger than that for Eu(III). These results suggest that, although both 12 and 3a are surface active, 12 extracts metal ions at the interface more rapidly than 3a does, in agreement with the extraction results.
Finally, to explain why precipitate formation was observed in the extraction experiments with 13 and not with 12 and 14, we compared the calculated logP values of the three ligands (ESI†). Ligand 13 is predicted to be significantly less lipophilic than ligands 12 and 14, due to the presence of the ether oxygen atoms in ligand 13 that are absent in ligands 12 and 14.
In summary, we present a new route to amide-functionalized, camphor-derived ligands for selective actinide extraction. The ligands show high solubilities in 1-octanol, are able to extract and separate Am(III) and Cm(III) from the lanthanides with good selectivity, and exhibit rapid rates of metal extraction. However, the ligands are unable to separate Am(III) from Cm(III). In contrast to the unfunctionalized ligands 4, 17 and 18, two of the ligands do not form precipitates in contact with nitric acid solutions. We conclude that precipitate formation by camphor-derived ligands is influenced by the interplay between ligand hydrophobicity and ligand basicity, and precipitate formation can be avoided by optimizing ligand design. Further studies are underway on the evaluation of a broader library of these ligands.
We thank the EPSRC (EP/P004873/1) and Northumbria University for funding this research, and the Royal Society of Chemistry for awarding a Researcher Mobility Grant (R.B.). We also thank the EPSRC UK National Mass Spectrometry Facility at Swansea University for recording high-resolution mass spectra.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. L. Nash, C. Madic, J. Mathur and J. Lacquement, The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer; Dordrecht, 2011, vol. 4, pp. 2622–2798 Search PubMed.
- J. Magill, V. Berthou, D. Haas, J. Galy, R. Schenkel, H.-W. Wiese, G. Heusener, J. Tommasi and G. Youinou, Nucl. Energy, 2003, 42, 263–277 CAS.
- Fuel Cycle Stewardship in a Nuclear Renaissance, The Royal Society Science Policy Centre Report 10/11, The Royal Society, London, 2011 (ISBN: 978-0-85403-891-6).
- N. Kaltsoyannis and S. T. Liddle, Chemistry, 2016, 1, 659–662 CrossRef CAS.
- (a) G. Modolo, A. Wilden, A. Geist, D. Magnusson and R. Malmbeck, Radiochim. Acta, 2012, 100, 715–725 CrossRef CAS; (b) A. Geist, R. Taylor, C. Ekberg, P. Guilbaud, G. Modolo and S. Bourg, Proc. Chem., 2016, 21, 218–222 CrossRef; (c) A. Baker, A. Fells, M. J. Carrott, C. J. Maher and B. C. Hanson, Chem. Soc. Rev., 2022, 51, 3964–3999 RSC.
- For reviews, see: (a) Z. Kolarik, Chem. Rev., 2008, 108, 4208–4252 CrossRef CAS PubMed; (b) F. W. Lewis, M. J. Hudson and L. M. Harwood, Synlett, 2011, 2609–2632 CAS; (c) M. J. Hudson, L. M. Harwood, D. M. Laventine and F. W. Lewis, Inorg. Chem., 2013, 52, 3414–3428 CrossRef CAS PubMed; (d) P. J. Panak and A. Geist, Chem. Rev., 2013, 113, 1199–1236 CrossRef CAS PubMed; (e) A. Leoncini, J. Huskens and W. Verboom, Chem. Soc. Rev., 2017, 46, 7229–7273 RSC; (f) A. Geist and P. J. Panak, Solvent Extr. Ion Exch., 2021, 39, 128–151 CrossRef CAS.
- For recent examples, see: (a) C.-L. Xiao, C.-Z. Wang, L.-Y. Yuan, B. Li, H. He, S. Wang, Y.-L. Zhao, Z.-F. Chai and W.-Q. Shi, Inorg. Chem., 2014, 53, 1712–1720 CrossRef CAS PubMed; (b) X.-H. Kong, Q.-Y. Wu, J.-H. Lan, C.-Z. Wang, Z.-F. Chai, C.-M. Nie and W.-Q. Shi, Inorg. Chem., 2018, 57, 14810–14820 CrossRef CAS PubMed.
- (a) M. J. Hudson, C. E. Boucher, D. Braekers, J. F. Desreux, M. G. B. Drew, M. R. S. J. Foreman, L. M. Harwood, C. Hill, C. Madic, F. Marken and T. G. A. Youngs, New J. Chem., 2006, 30, 1171–1183 RSC; (b) M. R. S. Foreman, M. J. Hudson, M. G. B. Drew, C. Hill and C. Madic, Dalton Trans., 2006, 1645–1653 RSC; (c) F. W. Lewis, L. M. Harwood, M. J. Hudson, M. G. B. Drew, J. F. Desreux, G. Vidick, N. Bouslimani, G. Modolo, A. Wilden, M. Sypula, T.-H. Vu and J.-P. Simonin, J. Am. Chem. Soc., 2011, 133, 13093–13102 CrossRef CAS PubMed.
- S. Trumm, A. Geist, P. J. Panak and T. Fanghänel, Solvent Extr. Ion Exch., 2011, 29, 213–229 CrossRef CAS.
- (a) T. Money, Nat. Prod. Rep., 1985, 2, 253–289 RSC; (b) E. A. Shokova, J. K. Kim and V. V. Kovalev, Russ. J. Org. Chem., 2016, 52, 459–488 CrossRef CAS.
- Ligand 4 derived from enantiomerically pure (+)- or (–)-camphorquinone gives higher distribution ratios than ligand 4 derived from racemic camphorquinone, see: K. N. Tevepaugh, J. D. Carrick, S. Tai, J. G. Coonce, L. H. Delmau and D. D. Ensor, Solvent Extr. Ion Exch., 2016, 34, 13–25 CrossRef CAS.
- U. Huynh, S. L. McDonald, D. Lim, Md. N. Uddin, S. E. Wengryniuk, S. Dey and D. M. Coltart, J. Org. Chem., 2018, 83, 12951–12964 CrossRef CAS PubMed.
- (a) T. Poloński, J. Chem. Soc., Perkin Trans. 1, 1983, 305–309 RSC; (b) A. García Martínez, E. Teso Vilar, A. García Fraile and P. Martínez-Ruiz, Tetrahedron: Asymmetry, 2005, 16, 2329–2332 CrossRef.
- D. M. Laventine, A. Afsar, M. J. Hudson and L. M. Harwood, Heterocycles, 2012, 86, 1419–1429 CrossRef CAS.
- A. Geist, C. Hill, G. Modolo, M. R. S. J. Foreman, M. Weigl, K. Gompper, M. J. Hudson and C. Madic, Solvent Extr. Ion Exch., 2006, 24, 463–483 CrossRef CAS.
- A. V. Zaytsev, R. Bulmer, V. N. Kozhevnikov, M. Sims, G. Modolo, A. Wilden, P. G. Waddell, A. Geist, P. J. Panak, P. Wessling and F. W. Lewis, Chem. – Eur. J., 2020, 26, 428–437 CrossRef CAS PubMed.
- F. W. Lewis, L. M. Harwood, M. J. Hudson, M. G. B. Drew, V. Hubscher-Bruder, V. Videva, F. Arnaud-Neu, K. Stamberg and S. Vyas, Inorg. Chem., 2013, 52, 4993–5005 CrossRef CAS PubMed.
- S. Kobayashi, S. Nagayama and T. Busujima, J. Am. Chem. Soc., 1998, 120, 8287–8288 CrossRef CAS.
- See for example: (a) M. Steppert, I. Císařová, T. Fanghänel, A. Geist, P. Lindqvist-Reis, P. Panak, P. Štěpnička, S. Trumm and C. Walther, Inorg. Chem., 2012, 51, 591–600 CrossRef CAS PubMed; (b) D. M. Whittaker, T. L. Griffiths, M. Helliwell, A. N. Swinburne, L. S. Natrajan, F. W. Lewis, L. M. Harwood, S. A. Parry and C. A. Sharrad, Inorg. Chem., 2013, 52, 3429–3444 CrossRef CAS PubMed.
- A. Bhattacharyya, P. M. Förster, D. B. Rego and K. R. Czerwinski, Eur. J. Inorg. Chem., 2016, 921–927 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR and mass spectra, solvent extraction data, NMR titrations with lanthanide nitrate salts, and kinetics and interfacial tension measurements. See DOI: https://doi.org/10.1039/d2cc03876e |
| This journal is © The Royal Society of Chemistry 2022 |