
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Linear conjugated polymer photocatalysts with various linker units for photocatalytic hydrogen evolution from water†
Lunjie
Liu
a,
Michał A.
Kochman
b,
Wei
Zhao
a,
Martijn A.
Zwijnenburg
 *b and
Reiner Sebastian
Sprick
*c
*b and
Reiner Sebastian
Sprick
*c
aDepartment of Chemistry and Materials Innovation Factory, University of Liverpool, 51 Oxford Street, Liverpool, L7 3NY, UK
bDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: m.zwijnenburg@ucl.ac.uk
cDepartment of Pure and Applied Chemistry, University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: sebastian.sprick@strath.ac.uk
First published on 22nd August 2022
Abstract
Polymer photocatalysts have shown potential for light-driven hydrogen evolution from water. Here we studied the relative importance of the linker type in two series of conjugated polymers based on dibenzo[b,d]thiophene sulfone and dimethyl-9H-fluorene. The alkenyl-linked polymers were found to be more active photocatalysts than their alkyl and alkyne-linked counterparts. The co-polymer of dibenzo[b,d]thiophene sulfone and 1,2-diphenylethene has a hydrogen evolution rate of 3334 μmol g−1 h−1 and an external quantum efficiency of 5.6% at 420 nm.
Hydrogen is widely considered to have enormous potential to replace fossil fuels in the near future and will allow our societies to maintain sustainable development.1 Currently, most hydrogen is produced via steam reforming of methane,2 which produces large amounts of carbon dioxide, making it non-sustainable. For this reason, converting solar energy to hydrogen via photocatalytic water splitting with semiconductors has attracted much attention, in particular over the last decade, as it allows for the production of hydrogen without the emission of greenhouse gases.3 Although inorganic photocatalysts, such as TiO2 and other metal oxides, have been predominantly explored in this area,4,5 they are limited by low conversion efficiencies in the visible part of the solar spectrum.
Organic semiconductors, as photocatalysts for water splitting, offer potential advantages, such as low cost, tunability and the great structural diversity of possible building blocks.6,7 Inspired by the application of carbon nitride in this field,8 organic materials have been studied as photocatalysts for sacrificial hydrogen production, such as covalent organic frameworks (COFs),9,10 conjugated microporous polymers (CMPs),11,12 covalent triazine-based frameworks (CTFs),13,14 and linear polymers.15,16 Some organic materials even show good activity for overall water splitting (e.g., the linear conjugated polymer (P10) combined with BiVO4 in a Z-scheme17 or as a single light absorber with IrO2 as a co-catalyst,18 and crystalline carbon nitride with suitable co-catalysts19). To rationally design more candidate organic photocatalysts, it is important to understand the underlying structure-property-performance relationships. In our previous studies, we have demonstrated that introducing dibenzo[b,d]thiophene sulfone into polymers improves the hydrophilicity of the resulting material, which enhances the photocatalytic performance.10,15 In addition, the photocatalytic activity of polymer catalysts can be optimised by modifying the linkers. For example, extending the length of the linker in a CTF14 or COF9 results in increased photocatalytic hydrogen production performance. Besides, changing the elemental composition of the linker also induces an obvious effect on the photocatalytic performance.20 However, the strategy of varying the conjugation of the linker itself has not been studied systematically in linear polymers. So far, the only alkenyl-linked photocatalysts reported have been COFs.21,22
Herein, six conjugated linear polymers were synthesised via Pd(0)-catalysed Suzuki–Miyaura cross coupling polycondensation based on dibenzo[b,d]thiophene sulfone and dimethyl-9H-fluorene coupled with three different linkers: 1,2-diphenylethane, (E)-1,2-diphenylethene and 1,2-diphenylethyne (Fig. 1a, LS1-3 and LF1-3). The materials are obtained as insoluble powders from a precipitation reaction. When the materials were tested under visible illumination, it was found that alkenyl-linked LS2 exhibited the highest sacrificial hydrogen evolution rate (HER) under visible light, outperforming alkyne-linked LS1 and alkyl-linked LS3, with the same trends also being observed for the dimethyl-9H-fluorine-based materials.
 | ||
| Fig. 1 (a) Structures of linear conjugated polymers based on a dibenzo[b,d]thiophene sulfone unit (left, LS1–3) and dimethyl-9H-fluorene (right, LF1–3). (b) The solid-state UV-vis spectra of LS and LF polymers. | ||
The chemical structures of all polymers were studied by Fourier-transform infrared spectroscopy (FT-IR) (Fig. S1, ESI†). The LS polymers show a characteristic peak at 1160 cm−1, which can be assigned to stretching vibration of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO groups, which is not observed in the LF polymers, and the signal of C
SO groups, which is not observed in the LF polymers, and the signal of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C groups in LS3 and LF3 can be identified at around 2210 cm−1.23 The powder Xray diffraction (PXRD) patterns (Fig. S2, ESI†) indicated that the polymers were semi-crystalline materials, similar as observed in previous studies.24,25 The LS polymers appear to have a higher degree of long-range order as evident from their sharper diffraction peaks compared to LF polymers. Thermogravimetric analysis showed that the LF polymers were stable up to around 450 °C in N2, which was 50 °C higher than LS polymers under the same conditions (Fig. S3, ESI†). In both series, alkyne-linked polymers were more stable than alkenyl- and alkyl-linked polymers. The morphology of all the polymers was studied via scanning electron microscopy imaging (SEM, Fig. S4 and S5, ESI†). The LS polymers were mainly composed of flakes with various sizes, while the dominant morphologies of the LF polymers were agglomerated particles mixed with irregular blocks, which suggested that the host building blocks (dibenzo[b,d]thiophene sulfone moiety and the dimethyl-9H-fluorene moiety) had a significant effect on the morphology of the polymers, while the morphology was less affected by the linker. In addition, the Brunauer–Emmett–Teller surface areas (SABET) were calculated from N2 adsorption isotherms at 77 K. All polymers had low BET surface areas ranging from 26 m2 g−1 to 55 m2 g−1 (Table 1 and Fig. S6, ESI†) indicating that these materials are essentially non-porous as expected from previous work.24,25
C groups in LS3 and LF3 can be identified at around 2210 cm−1.23 The powder Xray diffraction (PXRD) patterns (Fig. S2, ESI†) indicated that the polymers were semi-crystalline materials, similar as observed in previous studies.24,25 The LS polymers appear to have a higher degree of long-range order as evident from their sharper diffraction peaks compared to LF polymers. Thermogravimetric analysis showed that the LF polymers were stable up to around 450 °C in N2, which was 50 °C higher than LS polymers under the same conditions (Fig. S3, ESI†). In both series, alkyne-linked polymers were more stable than alkenyl- and alkyl-linked polymers. The morphology of all the polymers was studied via scanning electron microscopy imaging (SEM, Fig. S4 and S5, ESI†). The LS polymers were mainly composed of flakes with various sizes, while the dominant morphologies of the LF polymers were agglomerated particles mixed with irregular blocks, which suggested that the host building blocks (dibenzo[b,d]thiophene sulfone moiety and the dimethyl-9H-fluorene moiety) had a significant effect on the morphology of the polymers, while the morphology was less affected by the linker. In addition, the Brunauer–Emmett–Teller surface areas (SABET) were calculated from N2 adsorption isotherms at 77 K. All polymers had low BET surface areas ranging from 26 m2 g−1 to 55 m2 g−1 (Table 1 and Fig. S6, ESI†) indicating that these materials are essentially non-porous as expected from previous work.24,25
| Polymer | IPa/V | EAa/V | Fundamental gapa/(e)V | Optical gapb/eV | SA BET /m2 g−1 | Transmissiond /% | Contact anglee/° | τ avg /ns | H2 Evolution rateg /μmol g−1 h−1 |
|---|---|---|---|---|---|---|---|---|---|
a IP and EA were predicted using B3LYP calculations, and the fundamental gap (Ef) was calculated by Ef = IP–EA.
b The optical gap was estimated by applying the Kubelka–Munk formula (αhν)^1/2 α Eg to the measured UV-vis reflection spectrum.
c Apparent BET surface areas were calculated from the N2 adsorption isotherms.
d Measured in H2O/MeOH/TEA.
e Measured as pellets against water.
f Estimated fluorescence lifetimes were obtained from fitting time-correlated single photon counting decays to a sum of three exponentials in water.
g Polymer photocatalyst (25 mg) in H2O/MeOH/TEA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, 25 mL) solution was irradiated by a 300 W Xe light source equipped with a λ > 420 nm filter. :1:1, 25 mL) solution was irradiated by a 300 W Xe light source equipped with a λ > 420 nm filter.
|
|||||||||
| LS1 | 1.26 | −2.03 | 3.29 | 2.63 | 53 | 0.5 | 65.4 (±0.6) | 1.43 | 1013 (±22) |
| LS2 | 0.87 | −1.79 | 2.66 | 2.50 | 52 | 0.3 | 56.3 (±0.8) | 1.57 | 3334 (±360) |
| LS3 | 1.08 | −1.77 | 2.85 | 2.54 | 29 | 0.6 | 59.0 (±0.6) | 1.04 | 1980 (±182) |
| LF1 | 0.81 | −2.53 | 3.34 | 2.73 | 53 | 27.3 | 69.4 (±0.8) | 0.30 | 40(±25) |
| LF2 | 0.62 | −2.15 | 2.77 | 2.61 | 26 | 24.3 | 59.4 (±0.5) | 0.13 | 539 (±115) |
| LF3 | 0.78 | −2.14 | 2.93 | 2.66 | 46 | 30.7 | 62.6 (±0.3) | 0.19 | 303 (±150) |
UV-vis diffuse reflectance spectra were recorded in the solid state to study the optical properties of the polymers. As shown in Fig. 1b, the spectra of the LS polymers are red-shifted relative to the LF polymers with the same linker and hence absorb more of the visible spectrum. In addition, the optical gap (Eg) was estimated via the Kubelka–Munk formula from the measured UV-vis spectra. For LS1–LS3, their optical gaps were estimated to be 2.63 eV, 2.50 eV and 2.54 eV, respectively, which is slightly smaller than those of their corresponding LF polymers (Table 1 and Table S1, ESI†). The electron affinity (EA) and ionisation potential (IP), as well as their excited state equivalents (IP* and EA*), were predicted using (TD-)DFT calculations (Fig. 2 and Table 1). These calculations followed a previously developed approach26,27 based on ΔDFT calculations using the B3LYP density functional28–30 in the presence of a dielectric continuum model (εr 80.1) to describe polymer strands near the polymer particle-water interface. EA/IP* governs the driving force for water reduction, while IP/EA* controls the driving force for water oxidation (or TEA oxidation in this case; Table 1). All of the polymers are predicted to have EA/IP* values that are more negative than the proton reduction potential at pH 12.3, the pH of the reaction mixture,31 indicating that all of them are thermodynamically able to reduce protons to hydrogen. Similarly, all polymers barring perhaps LF2 have IP/EA*values that are sufficiently positive to not only drive the overall oxidation of TEA but also the intermediate one-hole oxidation of TEA.
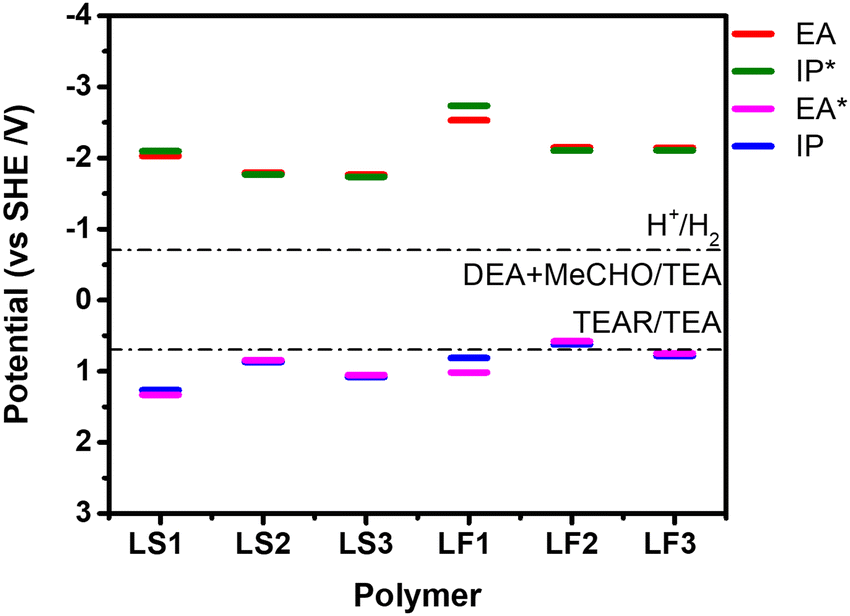 | ||
| Fig. 2 B3LYP-predicted EA, IP, EA*, and IP* potentials of LS1-3 and LF1-3. Solution half reactions are shown for pH 12.3, the measured pH of the H2O/MeOH/TEA mixture (MeCHO; acetaldehyde; TEAR; deprotonated TEA radical). | ||
The photocatalytic hydrogen production performance of powders of the polymers was studied as dispersions in a H2O/methanol (MeOH)/triethylamine (TEA) mixture (1:1:1) under visible light irradiation (λ > 420 nm, a 300 W Xe light source). Similar to other studies,25,32 we found that methanol does not act as a sacrificial donor; instead, it suppresses phase separation between water and the triethylamine (Fig. S7, ESI†). No additional co-catalysts were used; however, it should be noted that residual palladium originating from the palladium catalyst used in the polymer synthesis has been shown to act as a co-catalyst during hydrogen evolution.33 The palladium content of the materials in this study varied from 0.31% for LF2 to 0.75% for LF1 as determined by inductively coupled plasma/optical emission spectrometry. While this could contribute to differences in activity, it has been shown that when the residual Pd concentration was over a certain threshold, it only has a negligible additional effect on the photocatalytic performance.11,34 As shown in Fig. 3a, the HER of LS2 was 3334 μmol g−1 h−1, which was 1.7 times higher than that of LS3 and 3.3 times higher than that of LS1. For LF polymers, it was also found that the HER of LF2 was higher than those of the other two polymers, although the rate was only around one-fifth that of LS2 (539 μmol g−1 h−1). Therefore, in terms of photocatalytic performance, alkenyl-linked polymers outperformed the alkyne-linked and alkyl-linked polymers in this study. One remarkable observation is the fact that the alkyl-linked polymers show significant photocatalytic activity despite their lack of extended conjugation along the backbone. Similar observations have been made previously for dibenzo[b,d] thiophene sulfone oligomers,35 which also showed significant activity despite having limited conjugation by the mere fact of their lengths.
 | ||
| Fig. 3 (a) Time courses of photocatalytic H2 evolution of all photocatalysts made in this study; (b) extended photocatalysis run of LS2 over five 5-hour cycles. Experimental conditions: 25 mg of the polymer photocatalyst in a H2O/MeOH/TEA (1:1:1, 25 mL) solution was irradiated by a 300 W Xe light source with a λ > 420 nm cut-off filter. | ||
To evaluate the photostability of LS2, recycling experiments were performed over 25 hours under visible light illumination (λ > 420 nm) and as shown in Fig. 3b, no obvious change of activity was observed over this timeframe. Furthermore, no obvious changes in the morphology, FT-IR spectra and PXRD patterns of LS2 when comparing the as made material with the material after photocatalysis (Fig. S9 and S10, ESI†) were observed, indicating that LS2 has a good stability under operating conditions. External quantum efficiencies (EQEs) were measured at different wavelengths to further evaluate the performance of LS2. The highest EQE value of 5.6% was obtained at 420 nm, which is comparable to some other reported organic materials (Table S2, ESI†), such as phenyl-benzothiadiazole copolymer (Pt/B-BT-1,4, 4.01% at 420 nm),36 bipyridyl-based porous conjugated polymer (PCP4e, 1.8% at 350 nm),37 although still lower than the state of the art in organic polymer photocatalysts, for example, a donor–π–acceptor (D–π–A) conjugated organic copolymer (PyBS-3, 29.3% at 420 nm)38 and dibenzo[b,d]thiophene sulfone-dibenzo[b,d]thiophene co-polymer (P64, 20.7% at 420 nm).39 In addition, the measured EQEs of LS2 also matched the UV-vis absorption spectrum well (Fig. S11, ESI†), demonstrating that the hydrogen production is indeed a light-driven process.14
The wettability of the polymer powders was studied via contact angle measurements of pellets of the materials against water (Table 1 and Fig. S12, ESI†), fitted with a Young–Laplace model. It was found that alkenyl-linked polymers (56.3° for LS2 and 59.4° for LF2) have smaller contact angles than alkyne-linked polymers and alkyl-linked polymers. The dispersibility of the polymer photocatalyst powders was studied via turbidity measurements in the H2O/MeOH/TEA mixture reaction solution. Low transmission values indicate good dispersibility, while high values indicate that the material either floats up or settles at the bottom during the measurement. LS1–LS3 showed very similar and low transmission value (0.3–0.6%, Table 1), indicating that the powders of LS polymers disperse well in the reaction mixture. The powders of LF polymers in contrast had much higher transmission values (27.3%, 24.3% and 30.7% for LF1, LF2 and LF3, respectively). The better dispersibility of the LS polymers can be attributed to the increased hydrophilicity of materials that bear sulfone groups.15,38 The particle sizes of all the polymers were measured by static light scattering experiments (Table S3 and Fig. S13, ESI†). The median particle sizes of LS polymers ranged from 11.4 μm to 15.1 μm and were smaller than those of the LF polymers (18.6 μm to 20.3 μm). The photoluminescence spectra were also measured (Fig. S14, ESI†), an obvious redshift can be observed comparing alkyl-linked polymers to alkenyl- and alkyne-linked polymers, which can be explained by the change of optical gap induced by the variation of the linkers.11 In addition, time-correlated single photon counting (TCSPC) showed that LS2 had the longest fluorescence lifetime of 1.57 ns among these polymers, and the fluorescence lifetimes of LS polymers were longer than those of LF polymers (Table 1, Table S4 and Fig. S15–S17, ESI†). In general, longer exciton lifetimes are considered to increase photocatalytic hydrogen evolution performance; however, LF2 showed the shortest lifetime, while the HER activity was the highest in the LF series. Similar observations have been made previously,23,39 showing limitations of the technique, as non-emissive states that could be responsible for hydrogen production are not studied with TCSPC. When correlating the HER with various material properties (Fig. S18 and S19, ESI†), it was found that no single property dominated the HER performance, as reported previously.23,39 Having said this, it appears that the IP positions, dispersibility, hydrophilicity and optical gaps have the largest effect on the materials’ activity, with other factors such as exciton separation efficiency also potentially being important.
In summary, we synthesized two series of linear conjugated polymers and compared the effect of the linker on the photocatalytic activity for sacrificial hydrogen evolution from water. It was found that alkenyl-linked polymers are more active compared to alkyne-linked polymers with alkyl-linked polymers being the least active. These changes in the activities can be mainly attributed to the changes in the visible light absorption and hydrophilicity of the materials. Polymer LS2, the best material in this study showed high HER of 3334 μmol g−1 h−1 with an EQE of 5.6% at 420 nm. Remarkably, even though the activity of alkyl-linked polymers is low they nevertheless show significant activity despite the lack of extended conjugation.
We thank the Engineering and Physical Sciences Research Council (EPSRC) for financial support under Grant EP/N004884/1. L. L. thanks the China Scholarship Council for a PhD studentship. R. S. S thanks the University of Strathclyde for financial support through The Strathclyde Chancellor's Fellowship Scheme.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. H. Kim, D. Hansora, P. Sharma, J.-W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- A. M. Abdalla, S. Hossain, O. B. Nisfindy, A. T. Azad, M. Dawood and A. K. Azad, Energy Convers. Manage., 2018, 165, 602–627 CrossRef CAS.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- M. Z. Rahman, M. G. Kibria and C. B. Mullins, Chem. Soc. Rev., 2020, 49, 1887–1931 RSC.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- X. Wang, K. Msprickaeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomäcker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- Z. Wang, X. Yang, T. Yang, Y. Zhao, F. Wang, Y. Chen, J. H. Zeng, C. Yan, F. Huang and J.-X. Jiang, ACS Catal., 2018, 8, 8590–8596 CrossRef CAS.
- J. Xie, S. A. Shevlin, Q. Ruan, S. J. A. Moniz, Y. Liu, X. Liu, Y. Li, C. C. Lau, Z. X. Guo and J. Tang, Energy Environ. Sci., 2018, 11, 1617–1624 RSC.
- C. B. Meier, R. S. Sprick, A. Monti, P. Guiglion, J.-S. M. Lee, M. A. Zwijnenburg and A. I. Cooper, Polymer, 2017, 126, 283–290 CrossRef CAS.
- R. S. Sprick, K. J. Cheetham, Y. Bai, J. Alves Fernandes, M. Barnes, J. W. Bradley and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 7125–7129 RSC.
- X.-H. Zhang, X.-P. Wang, J. Xiao, S.-Y. Wang, D.-K. Huang, X. Ding, Y.-G. Xiang and H. Chen, J. Catal., 2017, 350, 64–71 CrossRef CAS.
- Y. Bai, K. Nakagawa, A. J. Cowan, C. M. Aitchison, Y. Yamaguchi, M. A. Zwijnenburg, A. Kudo, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 16283–16290 RSC.
- Y. Bai, C. Li, L. Liu, Y. Yamaguchi, M. Bahri, H. Yang, A. Gardner, M. A. Zwijnenburg, N. D. Browning, A. J. Cowan, A. Kudo, A. I. Cooper and R. S. Sprick, Angew. Chem., Int. Ed., 2022 DOI:10.1002/anie.202201299.
- L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin, Z. Yu and X. Wang, Nat. Catal., 2020, 3, 649–655 CrossRef CAS.
- Y. Wang, M. K. Bayazit, S. J. A. Moniz, Q. Ruan, C. C. Lau, N. Martsinovich and J. Tang, Energy Environ. Sci., 2017, 10, 1643–1651 RSC.
- C. Mo, M. Yang, F. Sun, J. Jian, L. Zhong, Z. Fang, J. Feng and D. Yu, Adv. Sci., 2020, 7, 1902988 CrossRef CAS PubMed.
- J. Kang, S. Huang, K. Jiang, C. Lu, Z. Chen, J. Zhu, C. Yang, A. Ciesielski, F. Qiu and X. Zhuang, Adv. Funct. Mater., 2020, 30, 2000857 CrossRef CAS.
- L. Liu, M. A. Kochman, Y. Xu, M. A. Zwijnenburg, A. I. Cooper and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 17242–17248 RSC.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- P. Guiglion, C. Butchosa and M. A. Zwijnenburg, J. Mater. Chem. A, 2014, 2, 11996–12004 RSC.
- P. Guiglion, A. Monti and M. A. Zwijnenburg, J. Phys. Chem. C, 2017, 121, 1498–1506 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- Y. Bai, L. Wilbraham, H. Gao, R. Clowes, H. Yang, M. A. Zwijnenburg, A. I. Cooper and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 19958–19964 RSC.
- R. S. Sprick, B. Bonillo, M. Sachs, R. Clowes, J. R. Durrant, D. J. Adams and A. I. Cooper, Chem. Commun., 2016, 52, 10008–10011 RSC.
- J. Kosco and I. McCulloch, ACS Energy Lett., 2018, 3, 2846–2850 CrossRef CAS.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- C. M. Aitchison, M. Sachs, M. A. Little, L. Wilbraham, N. J. Brownbill, C. M. Kane, F. Blanc, M. A. Zwijnenburg, J. R. Durrant, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 8744–8756 RSC.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS PubMed.
- L. Li, Z. Cai, Q. Wu, W.-Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed.
- C. Shu, C. Han, X. Yang, C. Zhang, Y. Chen, S. Ren, F. Wang, F. Huang and J.-X. Jiang, Adv. Mater., 2021, 33, 2008498 CrossRef CAS PubMed.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc03810b |
| This journal is © The Royal Society of Chemistry 2022 |