
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcidochromism of donor–acceptor Stenhouse adducts in organic solvent†
Antonio
Fiorentino‡
a,
Brian
Sachini‡
b,
Stefano
Corra
 bc,
Alberto
Credi
bc,
Cristina
Femoni
c,
Aurore
Fraix
d and
Serena
Silvi
*ab
bc,
Alberto
Credi
bc,
Cristina
Femoni
c,
Aurore
Fraix
d and
Serena
Silvi
*ab
aDipartimento di Chimica “G. Ciamician”, Università di Bologna, via Selmi 2, 40126, Bologna, Italy. E-mail: serena.silvi@unibo.it
bCLAN-Center for Light Activated Nanostructures, Istituto ISOF-CNR, via Gobetti 101, 40129, Bologna, Italy
cDipartimento di Chimica Industriale “Toso Montanari”, Università di Bologna, Viale del Risorgimento 4, 40136, Bologna, Italy
dPhotoChemLab, Department of Drug and Health Sciences, University of Catania, 95125, Catania, Italy
First published on 15th August 2022
Abstract
First generation DASA derivatives can be reversibly isomerized from the coloured, open form to the colourless, closed isomer upon protonation, thus behaving as acidochromic compounds in halogenated organic solvent.
Donor–acceptor Stenhouse adducts (DASAs) are a new class of photochromic compounds,1,2 which can be synthesized straightforwardly from cheap and commercially available starting materials.3,4 Additionally, these compounds exhibit negative photochromism, coupled with a dramatic change in the polarity of the molecule.5,6 In fact, upon light irradiation or increasing the polarity of the medium the photochrome switches from an open coloured form to a closed, zwitterionic or neutral cyclopentenone colourless form. For these reasons, DASAs are becoming increasingly popular for several applications.7,8 Moreover, their highly modular structure allows the donor and/or acceptor extremities to be varied, tailoring the photochromic properties such as the absorption maximum, the equilibrium constant between the two forms, and the interconversion rates.6,9,10
The use of such photochromes, however, can be hampered by the complexity of their switching pathway.11 Moreover, in the open form, these molecules can exist in several configurations, depending on the surrounding medium which affects the double-bond configurations as well as keto–enol tautomerism.5,12
Here we report on the acid-induced cyclization13 of two first generation DASAs with a diethyl amine donor and either a barbituric acid acceptor 1 or a Meldrum acid acceptor 2 (Scheme 1) in dichloromethane solution. Despite their poor switching abilities, we have selected this particular class of molecules because they present an excellent selectivity for the open forms 1a and 2a in halogenated solvents,14 which simplifies the characterization of the species and the rationalization of the switching process. Moreover, given the complexity of the equilibria involved in the switching pathway, we selected the strong trifluoromethanesulfonic acid (TfOH), in the attempt to promote the protonation reactions and have clear-cut processes, in order to simplify the experimental outcome as well as the data treatment.
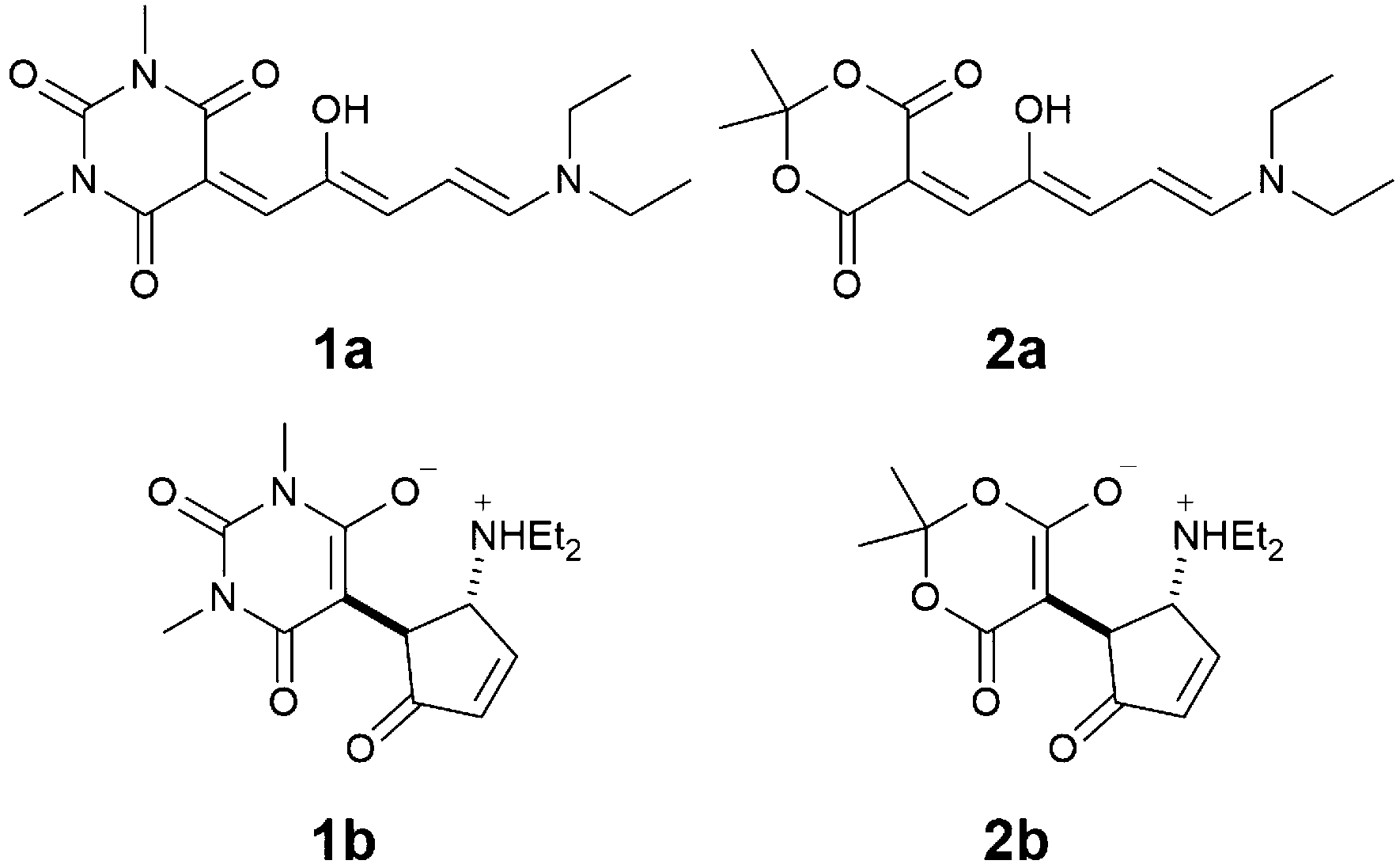 | ||
| Scheme 1 Molecular structures of DASA derivatives 1a and 2a and their cyclization products 1b and 2b. | ||
Compounds 1a and 2a were synthesized in two steps according to procedures reported in the literature (see ESI†).14
The absorption spectra of 1a and 2a in CH2Cl2 exhibit the characteristic intense and sharp bands in the visible, a weaker and broader band in the UV, as well as a weak emission (Table 1, Fig. 1 and ESI†).
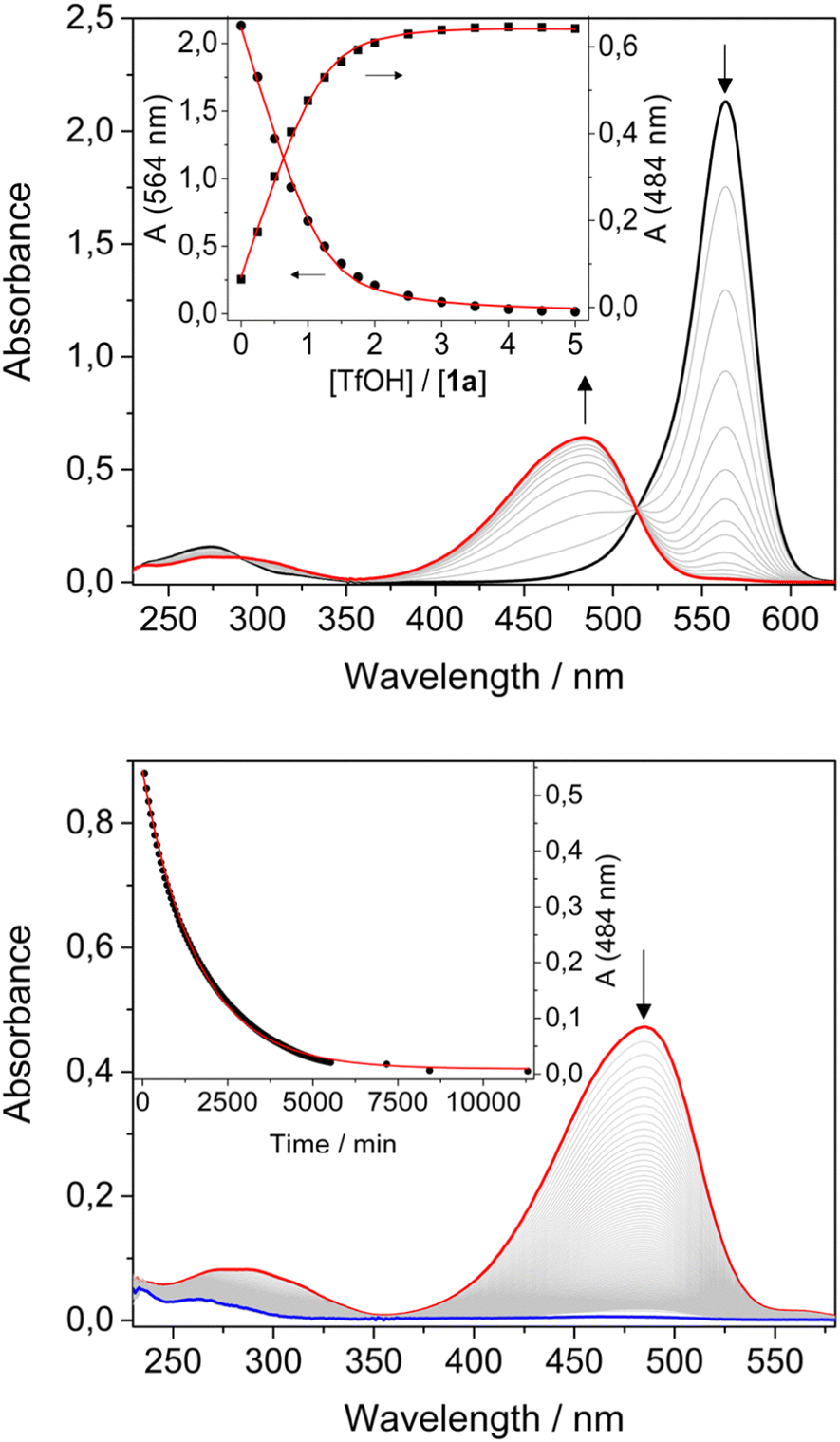 | ||
Fig. 1 Top: Absorption spectra of a 1.6 × 10−5 M solution of 1a upon addition of increasing amounts of TfOH; the inset shows the absorption changes at 484 nm and 564 nm as a function of acid added, the solid lines are the fitting curves of the experimental data according to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model. Bottom: Absorption spectra over time of a 1.5 × 10−5 M solution of 1a with 3 equivalents of acid added in the dark; the inset shows the absorption changes at 484 nm as a function of time, the solid line is the fitting curve of the experimental data according to a first order kinetic model. :1 binding model. Bottom: Absorption spectra over time of a 1.5 × 10−5 M solution of 1a with 3 equivalents of acid added in the dark; the inset shows the absorption changes at 484 nm as a function of time, the solid line is the fitting curve of the experimental data according to a first order kinetic model. | ||
Spectroscopic data confirmed that 1a and 2a are stable in their open form in CH2Cl2. Additionally, upon irradiation with visible light, no isomerization from the open to the closed form was observed in CH2Cl2, as expected considering the fast thermal isomerization reaction reported in the literature (t1/2 ≈ 11 s).14
Conversely, upon addition of TfOH, the colour of the solution changed from purple to orange/yellow. UV-Vis analysis for both compounds revealed a decrease of the absorption band in the visible region, accompanied by the formation of a new band at shorter wavelengths and by a red shift of the weak UV band (Table 1, Fig. 1a and ESI†). A blue shift and an increase of the luminescence quantum yield was also observed (Table 1). Titration with TfOH allowed to extract an equilibrium constant for a single protonation of 5 × 105 M−1 and 6.4 × 104 M−1 for 1a and 2a, respectively. The difference in the values of the equilibrium constants for the protonation reactions suggests an important role of the acceptor unit.
The two solutions of 1a and 2a were treated with an excess of TfOH, then left in the dark and absorption and emission spectra were recorded over time. The bands in the visible region disappeared while the bands in the UV decreased and shifted to higher energies. The fluorescence was quenched. The process is completed within 4 days for compound 1 and 12 hours for compound 2 (Fig. 1b and ESI†); the absorption changes can be fitted with a first order kinetic model, providing rate constants of 1 × 10−5 s−1 and 8 × 10−5 s−1, respectively.
These experiments suggest that the open forms of 1a and 2a can be quickly protonated, generating the open coloured protonated forms, 1a-H+ and 2a-H+. Depending on the medium the open isomer of DASAs can reside in a neutral or zwitterionic form.15 Protonation may, thus, occur either on the amine or on the enolate site of the acceptor moiety. The protonated open isomers then slowly cyclize to the corresponding protonated and non-conjugated closed forms 1b-H+ and 2b-H+.16 To confirm this hypothesis, structural characterization was sought by NMR spectroscopy. The 1H NMR spectra recorded in CD2Cl2 (5 mM) are consistent with DASAs 1a and 2a being the only species, as expected for first-generation DASAs in halogenated solvent (Fig. 2 and ESI†). Addition of 1 equivalent of TfOH to both 1a and 2a induced immediately large spectral variations. In particular, peak broadening was observed consistently with a fast protonation of the open-form DASAs (see the ESI†). Unfortunately, NMR titration of 1a with TfOH in CD2Cl2 to identify the protonation site did not provide a conclusive answer. The reaction was, then, allowed to proceed in the dark until complete transformation of 1a or 2a was observed.
 | ||
| Fig. 2 Stacked partial 1H NMR spectra (500 MHz, CD2Cl2, 298 K) of pristine 1a (a) and 1b-H+ formed after treatment of 1a 5 mM with one equivalent of TfOH 0.1 M (b). Resonance of proton H8 is highlighted in red. | ||
In both cases, the final 1H NMR spectrum displays sharp signals indicating the absence of any fast dynamic on the NMR timescale. The NMR structural characterization showed that the product of the reaction is a cyclized compound similar to that already reported for the photoisomerization.14 Interestingly, we observed the formation of an additional peak at 4.44 ppm for 1b-H+ and 4.89 ppm for 2b-H+. Such peaks have not been observed in light induced cyclization products of the two DASAs we investigated,3 suggesting that a new species is formed. Total correlation spectroscopy showed that the signal corresponds to the methyne proton (H8) of the 1,3-dicarbonyl moiety (Fig. 2b). These data suggest that, upon addition of acid, cyclization of the DASAs occurs similarly to changing the polarity of the medium. However, the presence of resonance H8 indicates that the keto tautomer is formed, rather than the enol tautomer,17 commonly observed upon photoisomerization in other media.
Additional confirmation of the structure of this species was sought by X-ray diffraction analysis. Single crystals were obtained by vapour diffusion of Et2O in a concentrated solution of 1a in CH2Cl2 with one equivalent of TfOH (Fig. 3). X-Ray diffraction revealed that the structure is in fact the cyclized form of the starting material (1b-H+). The distance between C12–C13 and C12–C17 is 1.53 Å and 1.48 Å respectively, which highlight a strong single bond character. Additionally, the bond lengths of the carbonyls C13–O18 and C17–O22 are both 1.19 Å, which demonstrate a strong double bond character. Similar observations were made for compound 2b-H+. These data are in line with compound 1b-H+ and 2b-H+ being diketo forms of the cyclized DASAs, as suggested by the NMR characterization.
 | ||
| Fig. 3 Single-crystal X-ray structure of compounds 1b-H+ (a) and 2b-H+ (b). Relevant hydrogen atoms are shown and their position was calculated and refined by a riding model. Trifluoromethanesulfonate counterions and all non-relevant hydrogen atoms were omitted for clarity. | ||
Overall, this structural characterization confirms that upon protonation of the open forms of the DASA derivatives in CH2Cl2, ring closing occurs to form the protonated closed isomers. The products are stable as ammonium salts with trifluoromethanesulfonate counterions in CH2Cl2, and exist as the diketo-tautomer of the acceptor moiety.
From a mechanistic point of view, ring closing requires isomerization of at least one double bond of the triene chain.11,18 Therefore, we envisioned that the isomerization reaction of the protonated open species could be accelerated in presence of light irradiation.19 Two solutions of 1a-H+ and 2a-H+ were, thus, irradiated in the visible region and the changes in the absorption spectra monitored over time. Spectral changes were consistent with those observed in the dark for the ring closing reaction. However, under light irradiation, the half-life of 1a-H+ was decreased up to three-fold, consistently with an acceleration of the reaction.
The effect of the concentration of acid on the cyclization kinetics was also investigated. For instance, compound 1a was cyclized in the presence of either 1 or 3 equivalents of acid. As expected from the protonation constant, different ratios between 1a and 1a-H+ were observed by UV-vis spectroscopy. Their evolution in time to form 1b-H+ was followed by monitoring the decrease of the two bands at 564 nm (1a) and 484 nm (1a-H+) (see the ESI†). From these experiments three observations can be made: (i) the overall kinetics of the isomerization process, namely the disappearance of the bands in the visible region assigned to the open isomers, is faster with respect to the kinetics in presence of an excess of acid; (ii) the two bands at 564 nm and 484 nm decrease with different rates; (iii) at the end of the transformation both bands are bleached, consistently with complete conversion of 1a and 1a-H+ in 1b-H+. Similar considerations can be made for compound 2.
The latter observation indicates that, when protonated, 1 and 2 are more stable in the closed form than in the open one. In other words, 1b and 2b are more basic than 1a and 2a.
The dependence of the isomerization rate on the amount of acid suggests the involvement of other reaction pathways.11 In the simplest scenario, the reaction network reported in Scheme 2 could describe the main processes, that is, protonation (horizontal processes) and isomerization (vertical processes) reactions.
 | ||
| Scheme 2 Reaction network connecting deprotonated (left column), protonated (right column), open (top row) and closed (bottom row) forms of 1. The same scheme applies to compound 2. Conventionally the equilibrium constants are defined for the reactions read from left to right and from top to bottom. | ||
The reversibility of the protonation–cyclization reaction cascade was also tested on both compounds, providing similar outcomes. For instance, addition of one equivalent of phosphazene base P1 either to 1a-H+ or to 1b-H+ restores the initial spectrum of 1a with 95% and 85% reversibility, respectively. It is worth noting that, while the deprotonation of 1a-H+ is a fast process, addition of base to a solution of 1b-H+ results in slow spectral changes, which were ascribed to the isomerization of 1b to 1a. 1H NMR analysis of the compound formed in these reaction conditions confirmed our hypothesis (see the ESI†). Fitting of time-dependent concentration data to a first order kinetic model, provided rate constants of 0.05 s−1 and 0.003 s−1 for compounds 1 and 2, respectively.
The evolution of the system in presence of acid can be simulated on the basis of the reactions reported in Scheme 2, by considering the following: 1b is more basic than 1a, therefore the equilibrium constant of reaction (4) is higher than that of reaction (1), which is 5 × 105 M−1 (see above); both protonation reactions (1) and (4) are fast; reaction (2) is slow, as suggested by the kinetics in excess of acid; the equilibrium constant of reaction (3) is smaller than 10−3, as the closed isomer is not stable in CH2Cl2,14 and the isomerization rate constant of 1b to 1a is 0.05 s−1 (see above); the equilibrium constants must fulfil the principle of detailed balance. By using reasonable values for the equilibrium and rate constants (see the ESI†), it is possible to qualitatively reproduce the observed behaviour.
In conclusion, our results show that, in halogenated organic solvent, protonation of first-generation DASAs leads to cyclization to a cationic closed form. The closed isomer was characterized by mono and bidimensional NMR spectroscopy and single crystal X-ray analysis.
This study demonstrates the possibility to use first-generation DASA derivatives as acidochromic compounds in halogenated organic solvent, by formation of a metastable coloured open species, which then rearranges to a stable colourless closed isomer. Additionally, the isomerization reactions can be accelerated by irradiation with visible light, and both protonated compounds can be reversibly converted back to the non-protonated open form by addition of a strong base.
Moreover, our results also indicate that the acid-induced cyclization of first-generation DASA-based switches must be taken into account upon their integration in more complex (supramolecular) systems.2
Financial support from the EU (H2020 ITN grant “ArtMoMa” no. 860434) and the Ministero dell’Università e della Ricerca (PRIN 20173L7W8K and 201732PY3X) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.References
- H. Dürr and H. Bouas-Laurent, ed., Photochromism: Molecules and Systems, Elsevier, 2013 Search PubMed.
- L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044 RSC.
- (a) S. Helmy, F. A. Leibfarth, S. Oh, J. E. Poelma, C. J. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2014, 136, 8169 CrossRef CAS PubMed; (b) S. Helmy, S. Oh, F. A. Leibfarth, C. J. Hawker and J. Read de Alaniz, J. Org. Chem., 2014, 79, 11316 CrossRef CAS PubMed.
- M. M. Lerch, W. Szymański and B. L. Feringa, Chem. Soc. Rev., 2018, 47, 1910 RSC.
- N. Mallo, P. T. Brown, H. Iranmanesh, T. S. C. MacDonald, M. J. Teusner, J. B. Harper, G. E. Ball and J. E. Beves, Chem. Commun., 2016, 52, 13576 RSC.
- J. R. Hemmer, S. O. Poelma, N. Treat, Z. A. Page, N. D. Dolinski, Y. J. Diaz, W. Tomlinson, K. D. Clark, J. P. Hooper, C. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2016, 138, 13960 CrossRef CAS PubMed.
- R. F. A. Gomes, J. A. S. Coelho and C. A. M. Afonso, Chem. – Eur. J., 2018, 24, 9170 CrossRef CAS PubMed.
- (a) D. E. Nánási, A. Kunfi, A. Ábrahám, P. J. Mayer, J. Mihály, G. F. Samu, E. Kiss, M. Mohai and G. London, Langmuir, 2021, 37, 3057 CrossRef PubMed; (b) C. Tonnelé, B. Champagne, L. Muccioli and F. Castet, Phys. Chem. Chem. Phys., 2018, 20, 27658 RSC; (c) M. M. Sroda, J. Lee, Y. Kwon, F. Stricker, M. Park, M. T. Valentine and J. Read de Alaniz, ACS Appl. Polym. Mater., 2022, 4, 141 CrossRef CAS.
- J. R. Hemmer, Z. A. Page, K. D. Clark, F. Stricker, N. D. Dolinski, C. J. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2018, 140, 10425 CrossRef CAS PubMed.
- R. Berraud-Pache, E. Santamaría-Aranda, B. de Souza, G. Bistoni, F. Neese, D. Sampedro and R. Izsák, Chem. Sci., 2021, 12, 2916 RSC.
- (a) M. Di Donato, M. M. Lerch, A. Lapini, A. D. Laurent, A. Iagatti, L. Bussotti, S. P. Ihrig, M. Medved’, D. Jacquemin, W. Szymański, W. J. Buma, P. Foggi and B. L. Feringa, J. Am. Chem. Soc., 2017, 139, 15596 CrossRef CAS PubMed; (b) H. Zulfikri, M. A. J. Koenis, M. M. Lerch, M. Di Donato, W. Szymański, C. Filippi, B. L. Feringa and W. J. Buma, J. Am. Chem. Soc., 2019, 141, 7376 CrossRef CAS PubMed.
- M. M. Sroda, F. Striker, J. A. Peterson, A. Bernal and J. Read de Alaniz, Chem. – Eur. J., 2021, 27, 4183 CrossRef CAS PubMed.
- Y.-D. Cai, T.-Y. Chen, X. Q. Chen and X. Bao, Org. Lett., 2019, 21, 7445 CrossRef CAS PubMed.
- N. Mallo, E. D. Foley, H. Iranmanesh, A. D. W. Kennedy, E. T. Luis, J. Ho, J. B. Harper and J. E. Beves, Chem. Sci., 2018, 9, 8242 RSC.
- M. M. Sroda, F. Stricker, J. A. Peterson, A. Bernal and J. Read de Alaniz, Chem. – Eur. J., 2021, 27, 4183 CrossRef CAS PubMed.
- L. Kortekaas, J. Chen, D. Jacquemin and W. R. Browne, J. Phys. Chem. B, 2018, 122, 6423 CrossRef CAS PubMed.
- Y. Shpinov, A. Schlichter, P. Pelupessy, T. Le Saux, L. Jullien and B. Adelizzi, Chem. – Eur. J., 2022 DOI:10.1002/chem.202200497.
- C. Berton, D. M. Busiello, S. Zamuner, E. Solari, R. Scopelliti, F. Fadaei-Tirani, K. Severin and C. Pezzato, Chem. Sci., 2020, 11, 8457 RSC.
- Y. Liao, Acc. Chem. Res., 2017, 50(8), 1956 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2181185 and 2181186. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc03761k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |