
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding the versatility and scope of the oxime ligation: rapid bioconjugation to disulfide-rich peptides†
Anke
Hering‡
a,
Nayara
Braga Emidio‡
 a and
Markus
Muttenthaler
*ab
a and
Markus
Muttenthaler
*ab
aInstitute for Molecular Bioscience, The University of Queensland, Brisbane, 4072, Australia. E-mail: m.muttenthaler@uq.edu.au
bInstitute of Biological Chemistry, University of Vienna, Währingerstraße 38, Vienna, 1090, Austria. E-mail: markus.muttenthaler@univie.ac.at
First published on 26th July 2022
Abstract
The oxime ligation is a valuable bioorthogonal conjugation reaction but with limited compatibility with disulfide-rich peptides/proteins and time-sensitive applications. Here we overcome these limitations by introducing a strategy that supports regiospecific control, oxidative folding, production of stable aminooxy-precursors for on-demand modification, and complete ligation within 5 min.
The oxime ligation is a useful bioorthogonal reaction between a nucleophilic aminooxy group (H2N–O–R) and an electrophilic carbonyl group (e.g. aldehyde/ketone) (Fig. 1a).1,2 The reaction is typically carried out in aqueous media and catalysed by aniline or phenylenediamine derivates (Fig. 1b and c).2–7 It is a reliable and versatile conjugation technique due to the mild reaction conditions, the high chemoselectivity, and the hydrolytic stability of the oxime bond.8 It does not require metal ion catalysts, which can cause problems with purifications and certain classes of peptides/proteins.8–11 The oxime ligation has been successfully applied for the preparation of bioconjugates, including polymer-proteins,12,13 peptide dendrimers,14 oligonucleo-peptides,15,16 glycoconjugates,17 protein–protein probes,1818F-PET tracers19,20 and hydrogels.3,21
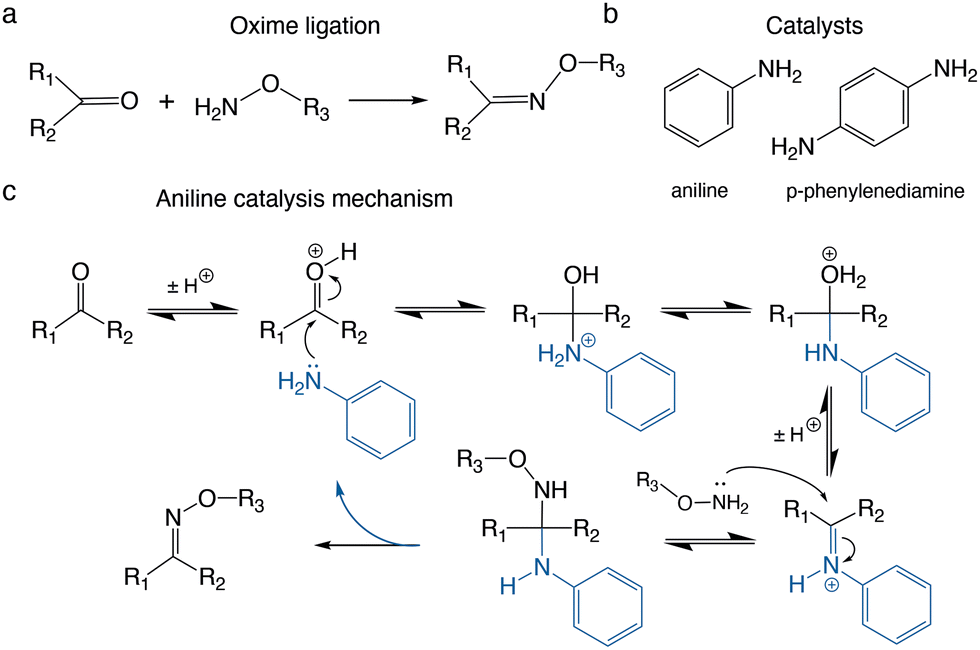 | ||
| Fig. 1 The oxime ligation and commonly used catalysts. (a) Reaction of an aminooxy and an aldehyde or ketone to form an oxime bond. (b) Aniline and p-phenylenediamine accelerate the reaction by acting as catalysts. (c) Nucleophilic catalysis mechanism of aniline (blue) during the oxime ligation. | ||
The synthesis of aminooxy-peptide precursors is challenging due to the high reactivity of the aminooxy moiety. Strategies have been developed to overcome this problem, including orthogonally protected aminooxy acetic acid (Aoa) derivates compatible with Fmoc- and Boc-SPPS (Fmoc-Aoa, Boc-Aoa, Boc2-Aoa).22 Boc2-Aoa was developed to avoid N-overacylation when using HCTU/DIPEA.22–24 Alternatively, preactivated NHS-ester, EEDQ (N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline) or Eei-Aoa (2-(1-ehoxyethylideneaminooxy)acetic acid) can be used.25,26
While this works for linear peptides, the aminooxy group is not compatible with the oxidative folding step for producing disulfide-rich peptides or proteins (Fig. S1a, ESI†). Indeed, the oxime ligation has only been reported for two disulfide-rich peptides, namely insulin and the c-Met peptide.27,28 In these studies, the ligation was carried out either before the folding step, or the aminooxy moiety was introduced after folding, circumventing the problem without solving it.27,28 Neither of these two strategies is ideal since they require two purification steps (after ligation and folding), which takes time and lowers the yields. Moreover, the introduction of the aminooxy group remains restricted to the N-terminus.
The handling and storing of aminooxy-containing peptides are also difficult due to the aminooxy's high reactivity towards aldehydes and ketones, including the laboratory solvent acetone. These limitations of an otherwise powerful, efficient, and clean bioorthogonal reaction prompted us to investigate new strategies to expand the scope of the oxime ligation.
We envisioned a strategy that could produce stable aminooxy precursors compatible with the folding of disulfide-rich peptides, long-term storage, and late-stage modifications. We furthermore realised that for time-sensitive applications such as the labelling with 18F (109.7 min half-life) to produce PET tracers,20,29 reaction conditions with vastly faster ligation kinetics are required than the currently used multi-hour protocols (Fig. 2a). 18F-labelling is typically carried out via an 18F-containing group compatible with different chemistries. Examples include activated carboxylic acids to react with free amines, maleimides to react with thiols, or alkynes/azides for click chemistry.20 These approaches have their own limitations, including lack of regioselective control when multiple free amines are present, and compatibility issues with disulfide-rich peptides/proteins for using maleimides or Cu-catalysts for click chemistry.20 The oxime ligation would be a powerful addition to the labelling repertoire if the reaction could be accelerated and made compatible with disulfide-rich peptides/proteins.
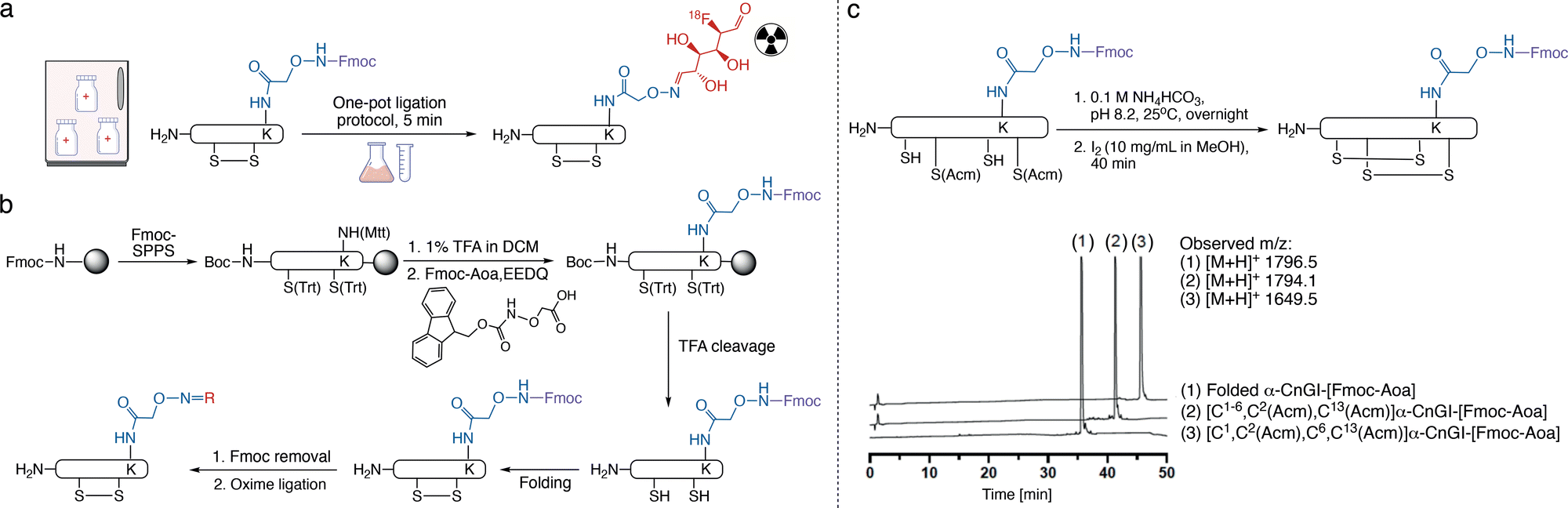 | ||
| Fig. 2 Expanding the scope of the oxime ligation. (a) Application scheme of the stable Fmoc-aminooxy-containing precursor peptide used with the rapid one-pot ligation protocol to support on-demand and on-site preparation of radioactive tracers for clinical settings. (b) Synthetic strategy for the regiospecific introduction of Fmoc-Aoa within a peptide during Fmoc-SPPS, followed by TFA cleavage and oxidative folding of the Fmoc-aminooxy-containing peptide, leading to a fully folded Fmoc-aminooxy-containing peptide precursor compatible with long-term storage. New functional groups can be conveniently introduced via Fmoc-removal and standard oxime ligation protocols. EEDQ: N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline. (c) Scheme of the directed disulfide bond formation of a two-disulfide-bond-containing peptide including the Fmoc-protected aminooxy group. Analytical C18-RP-HPLC traces of the directed folding of α-conotoxin analogue α-CnGI-[Fmoc-Aoa] with observed and calculated masses. Peak (1): reduced [C1,C2(Acm),C6,C13(Acm)]-α-CnGI-[Fmoc-Aoa]; peak (2): partially folded [C1–6,C2(Acm),C13(Acm)]-α-CnGI-[Fmoc-Aoa]; peak (3): fully folded [C1–6,C2–13]-α-CnGI-[Fmoc-Aoa]. α-CnGI sequence: CCHPACGKYFKC*, with *C-terminal amide and Fmoc-Aoa on K8. | ||
Thus, we first pursued regioselective on-resin incorporation of the aminooxy group into disulfide-bond-containing peptides (Fig. 2a). As a model peptide, we used the nonapeptide oxytocin (OT), which has a disulfide bond between Cys1 and Cys6. We replaced Leu8 with Lys(Mtt) (OTK8) as a controllable side chain handle to introduce the aminooxy group. Position 8 was selected based on previous studies demonstrating that position 8 modifications are well-tolerated in terms of bioactivity.30–32 OTK8 was assembled via Fmoc-SPPS on a Rink-amide resin, and Boc-Cys(Trt) was used as the N-terminal amino acid. The Mtt group was removed with 1% TFA in dichloromethane (DCM), and Fmoc-Aoa was coupled to the now unprotected ε-amino group of Lys8 using EEDQ as the coupling reagent. OTK8[Fmoc-Aoa] was cleaved from the resin using TFA and scavengers, with the aminooxy group remaining Fmoc-protected.
Exposure of reduced/linear OTK8[Fmoc-Aoa] to common oxidative folding conditions for 12 hours demonstrated that the Fmoc-protected aminooxy group remained intact, yielding folded OTK8[Fmoc-Aoa] in high purity (>90%) with no side product formation observed (Fig. S1b, ESI†). We then validated our strategy under conditions of directed disulfide bond formation using pairs of Cys(Acm) groups, one of the most commonly used directed folding strategies. We applied our strategy to α-CnIG (CCHPACGKYFKC*), an α-conotoxin from the venom of Conus consors, with disulfide bonds between Cys1–6 and Cys.2–13 α-Conotoxins are an important class of venom peptides from the marine predatory cone snail that potently and selectively inhibit nicotinic acetylcholine receptors.33,34 α-Conotoxins, like many other disulfide-rich peptides, are synthetically produced via a directed disulfide-bond formation strategy and are ideal models to demonstrate the compatibility of our aminooxy strategy with directed folding. α-CnIG was assembled by Fmoc-SPPS with the side chains of Cys1–6 and Cys2–13 protected with Trt and Acm, respectively. Boc-Cys1(Trt) was used as the N-terminal amino acid and Lys8(Mtt) as the aminooxy handle. After complete peptide assembly, the Mtt group was removed with 1% TFA/DCM, and Fmoc-Aoa was coupled to Lys8. Sidechain deprotection and peptide cleavage were carried out with TFA and scavengers, leaving Cys2/13 and Lys8-Aoa protected with Acm and Fmoc, respectively. The first disulfide bond (Cys1–6) was formed in 0.1 M NH4HCO3 at pH 8.2, followed by RP-HPLC purification. The second disulfide bond (Cys2–13) was then formed through I2 oxidation, followed by another purification (Fig. 2c and Fig. S2, ESI†). Both oxidations proceeded cleanly with no side reactions, confirming the compatibility with directed disulfide bond formation.
We then evaluated the long-term stability by monitoring OTK8[Fmoc-Aoa] dissolved in 0.1% TFA or 50% ACN/0.1% TFA over three months at 4 °C or as a lyophilised powder, confirming the high stability of the Fmoc-aminooxy-containing precursors (Fig. S3, ESI†).
This strategy therefore overcomes the first set of limitations, supporting regiospecific incorporation of the aminooxy moiety, compatibility with oxidative folding protocols and disulfide-rich peptides/proteins, and the preparation of stable Fmoc-aminooxy-containing precursors for long-term storage and late-stage modifications. The latter should be particularly useful for preparing peptide radiotracers as theranostics, a rapidly expanding field.35,36
Next, we aimed at accelerating the reaction kinetics. The oxime ligation with peptides and proteins is typically carried out over several hours in aqueous media at pH 4–5 with either aniline or p-phenylenediamine (pPDA) as catalysts (μM reactants, 10 mM catalysts).1,37,38 While these ligation times are acceptable for the majority of applications, they are too slow for radiochemistry applications that use 18F, which ideally requires complete ligation within minutes due to its short half-life (109.7 min). Ligation kinetics are driven by the concentration of the reactants, with solubility a recognised bottleneck. We therefore investigated the ligation kinetics in different solvent systems, including H2O, 80% acetonitrile (ACN), 80% ethanol (EtOH), and DMF, using purified OTK8[Fmoc-AoA] along with benzaldehyde, acetophenone or D-glucose as our model reactants, and pPDA as the catalyst.
First, we analysed the Fmoc removal kinetics using 30% (v/v) piperidine in these four solvents (Fig. S4, ESI†). Fmoc removal occurred within seconds in all tested solvents, and no racemisation was observed. The move to more organic solvents allowed higher ligand concentrations, which substantially accelerated reaction kinetics, with ligations of OTK8[AoA] with benzaldehyde and acetophenone completed in seconds compared to the slower and incomplete ligations in water (Fig. S5, ESI†). The reaction rate of D-glucose, however, even though faster than in water, was still not fast enough for rapid radiochemistry applications. The slower reaction kinetics are due to the energetic equilibrium between the cyclic and linear aldehyde conformation of D-glucose, with the cyclic conformation energetically favoured, but only the linear aldehyde conformation able to react with the aminooxy group (Fig. S5, ESI†).39,40 To overcome this problem, we increased the excess of D-glucose from 1 eq. to 100 eq. with 2 eq. pPDA as the catalyst in anhydrous DMF and the temperature to 75 °C. This improved the reaction rate but also induced unexpected dimerization of the peptide conjugate. This was resolved by replacing pPDA (two free amines) with aniline (single amine), resulting in clean ligation of OTK8[Aoa] with D-glucose within 5 min (compared to >1 h before optimisation). With the ligation reaction optimised, the next step was to develop a rapid one-pot Fmoc-removal, labelling and purification protocol to support time-sensitive applications. We dissolved OTK8[Fmoc-Aoa] in pre-heated 30% piperidine/anhydrous DMF (20 mM) for 1 min at 75 °C. The reaction was quenched with neat TFA (∼30% v/v) followed by the addition of pre-heated aniline (2 eq.) and D-glucose (100 eq.) in anhydrous DMF. The reaction was quenched after 5 min with acetone. Using this one-pot protocol >95% of OTK8[Aoa] reacted with D-glucose within 5 min (Fig. 3a). Fast C18-RP-HPLC purification (5–35% B in 15 min) yielded OTK8[Aoa-D-glucose] in high purity (>95%) (Fig. 3a).
 | ||
| Fig. 3 One-pot Fmoc-removal and oxime ligation of OTK8[Fmoc-Aoa] with D-glucose (a) or FDG (b). The Fmoc group of OTK8[Fmoc-AoA] was removed with 30% piperidine (1 min at 75 °C) and the reaction quenched with TFA (∼30% v/v). Aniline (2 eq.) and D-glucose (100 eq.) or pPDA (2 eq.) and FDG (100 eq.) were added, and the reaction mixed for 5 min at 75 °C. The reaction process was monitored by analytical C18 RP-HPLC and LC-MS. *, excess OTK8[Aoa] was quenched with acetone. Insets: Analytical C18-RP-HPLC traces and MS data of purified OTK8[Aoa-D-glucose] and OTK8[Aoa-FDG]. | ||
To apply our protocol to clinically more relevant material, we performed the ligation reaction also with fluorodeoxyglucose (FDG), a clinically used and readily available 18F source for PET imaging.20 Facile introduction of 18FDG into peptides via oxime ligation represents a new and highly efficient way of producing PET tracers that play an increasingly important role in oncology and other applications. Interestingly, the reaction conditions optimised for D-glucose were not sufficient for FDG ligation (>1 h for complete ligation). We however resolved this by changing the aniline catalyst back to pPDA and did not observe any dimerization as with D-glucose.
The final FDG labelling protocol entailed OTK8[Fmoc-Aoa] dissolution (20 mM) in pre-heated (75 °C) 30% piperidine/anhydrous DMF to remove the Fmoc group (1 min), which was quenched with neat TFA (∼30%, v/v). pPDA (2 eq.) and FDG (100 eq.) in anhydrous DMF were added, and the ligation reaction was quenched after 5 min with acetone (10% v/v). Analytical RP-HPLC and MS analysis confirmed complete product conversion within 5 min, and RP-HPLC purification yielded OTK8[Aoa-FDG] in >95% purity (Fig. 3b).
In summary, we developed a new strategy for regioselective control of the oxime ligation and compatibility with disulfide-rich peptides/proteins. The Fmoc-protected aminooxy group was stable to standard and directed oxidative folding conditions and Fmoc-aminooxy-containing precursors can be kept in long-term storage for on-demand bioconjugation and labelling. This is highly useful for preparing radiotracers, where adding the radionuclide is the final and time-sensitive step before injecting the tracer into patients. Our rapid one-pot labelling protocol enabled efficient 18F introduction into peptides/proteins within minutes, highlighting the application potential of this approach for (pre)clinical PET imaging.
Taken together, these new strategy and protocols considerably expand the versatility and scope of the oxime ligation, rendering it now an even more powerful tool that can be used with other bioconjugation reactions such as click chemistry, native chemical ligation, or the Staudinger reaction.41 This expands the chemical biology toolbox and creates new opportunities for late-stage modifications, radiotracer development, and biomaterial engineering.
A. H. and N. B. E. contributed equally to this work and conducted the experiments; M. M. conceived the idea and supervised the project; A. H., N. B. E and M. M. wrote the manuscript.
M. M. was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (714366) and by the Australian Research Council (ARC) (DE150100784, DP190101667 and FT210100266). We thank Prof. Paul Alewood (The University of Queensland) for supporting this work.
Conflicts of interest
There are no conflicts to declare.References
- K. Rose, J. Am. Chem. Soc., 1994, 116, 30–33 CrossRef CAS.
- S. M. Agten, P. E. Dawson and T. M. Hackeng, J. Pept. Sci., 2016, 22, 271–279 CrossRef CAS PubMed.
- G. N. Grover, J. Lam, T. H. Nguyen, T. Segura and H. D. Maynard, Biomacromolecules, 2012, 13, 3013–3017 CrossRef CAS PubMed.
- M. Rashidian, M. M. Mahmoodi, R. Shah, J. K. Dozier, C. R. Wagner and M. D. Distefano, Bioconjugate Chem., 2013, 24, 333–342 CrossRef CAS PubMed.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 118, 7743–7746 CrossRef.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- W. Tang and M. L. Becker, Chem. Soc. Rev., 2014, 43, 7013–7039 RSC.
- S. Li, H. Cai, J. He, H. Chen, S. Lam, T. Cai, Z. Zhu, S. J. Bark and C. Cai, Bioconjugate Chem., 2016, 27, 2315–2322 CrossRef CAS PubMed.
- N. W. Nairn, P. A. Bariola, T. J. Graddis, M. P. VanBrunt, A. Wang, G. Li and K. Grabstein, Bioconjugate Chem., 2015, 26, 2070–2075 CrossRef CAS PubMed.
- S. A. Fisher, A. E. G. Baker and M. S. Shoichet, J. Am. Chem. Soc., 2017, 139, 7416–7427 CrossRef CAS PubMed.
- T. L. Schlick, Z. B. Ding, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 3718–3723 CrossRef CAS PubMed.
- K. J. Mackenzie and M. B. Francis, J. Am. Chem. Soc., 2013, 245 Search PubMed.
- J. C. Breger, M. Muttenthaler, J. B. Delehanty, D. A. Thompson, E. Oh, K. Susumu, J. R. Deschamps, G. P. Anderson, L. D. Field, S. A. Walper, P. E. Dawson and I. L. Medintz, Nanoscale, 2017, 9, 10447–10464 RSC.
- N. Venkatesan and B. H. Kim, Chem. Rev., 2006, 106, 3712–3761 CrossRef CAS PubMed.
- Y. Singh, P. Murat and E. Defrancq, Chem. Soc. Rev., 2010, 39, 2054–2070 RSC.
- S. E. Cervigni, P. Dumy and M. Mutter, Angew. Chem., Int. Ed. Engl., 1996, 35, 1230–1232 CrossRef CAS.
- S. M. Agten, R. R. Koenen, H. Ippel, V. Eckardt, P. von Hundelshausen, K. H. Mayo, C. Weber and T. M. Hackeng, Angew. Chem., Int. Ed., 2016, 55, 14963–14966 CrossRef CAS PubMed.
- A. M. Senisik, C. Ichedef, A. Y. Kilcar, E. Ucar, K. Ari, D. Goksoy, Y. Parlak, B. E. S. Bilgin and S. Teksoz, J. Radioanal. Nucl. Chem., 2018, 316, 457–463 CrossRef CAS.
- X. G. Li, M. Haaparanta and O. Solin, J. Fluor. Chem., 2012, 143, 49–56 CrossRef CAS.
- J. G. Hardy, P. Lin and C. E. Schmidt, J. Biomater. Sci., Polym. Ed., 2015, 26, 143–161 CrossRef CAS PubMed.
- Y. Yang, in Side reactions in peptide synthesis, ed. Y. Yang, Academic Press, Oxford, 2016, pp. 235–256 Search PubMed.
- I. P. Decostaire, D. Lelièvre, H. Zhang and A. F. Delmas, Tetrahedron Lett., 2006, 47, 7057–7060 CrossRef CAS.
- J. Brask and K. J. Jensen, J. Pept. Sci., 2000, 6, 290–299 CrossRef CAS PubMed.
- V. Dulery, O. Renaudet and P. Dumy, Tetrahedron, 2007, 63, 11952–11958 CrossRef CAS.
- J. Brask and K. J. Jensen, J. Pept. Sci., 2000, 6, 290–299 CrossRef CAS PubMed.
- K. Thalluri, B. B. Kou, V. Gelfanov, J. P. Mayer, F. Liu and R. D. DiMarchi, Org. Lett., 2017, 19, 706–709 CrossRef CAS PubMed.
- R. B. Peter Brian Iveson, B. Indrevall and G. Getvoldsen, US Pat., US20130149241A1, 2011 Search PubMed.
- E. Miele, G. P. Spinelli, F. Tomao, A. Zullo, F. De Marinis, G. Pasciuti, L. Rossi, F. Zoratto and S. Tomao, J. Exp. Clin. Cancer Res., 2008, 27, 52 CrossRef PubMed.
- B. Chini, B. Mouillac, Y. Ala, M. N. Balestre, S. Trumpp-Kallmeyer, J. Hoflack, J. Elands, M. Hibert, M. Manning and S. Jard, et al. , EMBO J., 1995, 14, 2176–2182 CrossRef CAS PubMed.
- B. Chini, B. Mouillac, Y. Ala, M. N. Balestre, N. Cotte, S. Trumpp-Kallmeyer, J. Hoflack, J. Elands, M. Hibert and M. Manning, et al. , Adv. Exp. Med. Biol., 1995, 395, 321–328 CAS.
- B. Mouillac, B. Chini, M. N. Balestre, S. Jard, C. Barberis, M. Manning, E. Tribollet, S. Trumpp-Kallmeyer, J. Hoflack and J. Elands, et al. , Adv. Exp. Med. Biol., 1995, 395, 301–310 CAS.
- A. H. Jin, M. Muttenthaler, S. Dutertre, S. W. A. Himaya, Q. Kaas, D. J. Craik, R. J. Lewis and P. F. Alewood, Chem. Rev., 2019, 119, 11510–11549 CrossRef CAS PubMed.
- K. B. Akondi, M. Muttenthaler, S. Dutertre, Q. Kaas, D. J. Craik, R. J. Lewis and P. F. Alewood, Chem. Rev., 2014, 114, 5815–5847 CrossRef CAS PubMed.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- M. Fani, H. Maecke and S. Okarvi, Theranostics, 2012, 2, 481 CrossRef CAS PubMed.
- I. E. Decostaire, D. Lelievre, V. Aucagne and A. F. Delmas, Org. Biomol. Chem., 2014, 12, 5536–5543 RSC.
- M. Wendeler, L. Grinberg, X. Wang, P. E. Dawson and M. Baca, Bioconjugate Chem., 2014, 25, 93–101 CrossRef CAS PubMed.
- P. Finch and Z. Merchant, J. Chem. Soc., Perkin Trans. 1, 1975, 1682–1686 RSC.
- A. Mostad, Acta Chem. Scand., Ser. B, 1978, 32, 733–742 CrossRef.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures and material and methods (PDF). See DOI: https://doi.org/10.1039/d2cc03752a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |