
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nuclear Overhauser spectroscopy in hyperpolarized water – chemical vs. magnetic exchange†
Ludovica Martina
Epasto
 ab,
Philipp
Honegger
cd,
Kateryna
Che
a,
Fanny
Kozak
ab,
Florian
Jörg
bc,
Christian
Schröder
c and
Dennis
Kurzbach
*a
ab,
Philipp
Honegger
cd,
Kateryna
Che
a,
Fanny
Kozak
ab,
Florian
Jörg
bc,
Christian
Schröder
c and
Dennis
Kurzbach
*a
aUniversity of Vienna, Faculty of Chemistry, Department of Biological Chemistry, Währingerstr. 38, 1090 Vienna, Austria. E-mail: dennis.kurzbach@univie.ac.at
bUniversity of Vienna, Doctoral School in Chemistry (DoSChem), Währingerstr. 42, 1090 Vienna, Austria
cUniversity of Vienna, Faculty of Chemistry, Department of Computational Biological Chemistry, Währingerstr. 17, 1090 Vienna, Austria
dDepartment of Systems Biology, Harvard Medical School, 200 Longwood Avenue, Boston, MA 02115, USA
First published on 21st September 2022
Abstract
Dissolution dynamic nuclear polarization (dDNP) is a versatile hyperpolarization technique to boost signal intensities in nuclear magnetic resonance (NMR) spectroscopy. The possibility to dissolve biomolecules in a hyperpolarized aqueous buffer under mild conditions has recently widened the scope of NMR by dDNP. The water-to-target hyperpolarization transfer mechanisms remain yet unclear, not least due to an often-encountered dilemma of dDNP experiments: The strongly enhanced signal intensities are accompanied by limited structural information as data acquisition is restricted to short time series of only one-dimensional spectra or a single correlation spectrum. Tackling this challenge, we combine dDNP with molecular dynamics (MD) simulations and predictions of cross-relaxation rates to unravel the spin dynamics of magnetization flow in hyperpolarized solutions.
Spin hyperpolarization denotes a population distribution between nuclear spin levels far from thermal equilibrium. Such a state invokes strongly improved signal amplitudes, often over 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold enhanced, compared to conventional nuclear magnetic resonance (NMR) spectra. Various methods, from para hydrogenation to dynamic nuclear polarization (DNP) to optical pumping,1 enable the creation of hyperpolarized spin states for a wide array of applications. The use of hyperpolarized water to boost signal intensities in biomolecular NMR spectra has recently received ample attention2–5 as dissolving a target biomolecule in a hyperpolarized buffer enables new approaches at substantially enhanced sensitivity: residue- and time-resolved protein NMR,6 2D and 3D correlation spectra,7 protein folding and folding monitoring,8 membrane interactions,9 structural dynamics,10 and exchange processes.11 However, the understanding of the hyperpolarization transfer mechanism12 from the buffer to the targets remained incomplete; not least as data acquisition in the, most often used, dissolution DNP (dDNP) experiments is typically limited to short periods of 1–2 min that do not allow for determining the exchange pathways via complex pulse sequences. Important insights into hyperpolarization transfer by nuclear Overhauser effects (NOE) between water protons and molecules dissolved therein have, e.g., been achieved by Marco-Rius et al. (13C to 1H)13 or Hu et al. (1H to 1H or 19F).14 To this understanding, we here add a description of the interplay between direct solvent-solute NOE and exchange-relayed NOE in hyperpolarized water; an interesting phenomenon which invokes shifted weights of the different contributions to the magnetization flow as NOE and chemical exchange are differently affected by spin-hyperpolarization. Herein, we provide an understanding of these effects and, hence, also of the transient signal enhancements in biomolecular dDNP. In a dDNP experiment, the sample is ex situ pretreated to boost the spin resonances before transfer to an NMR spectrometer for detection. As the resulting spin-hyperpolarization after the transfer constitutes a non-equilibrium state, its lifetime is necessarily limited. We here demonstrate how to unravel the active transfer mechanisms despite a short time window by integrating dDNP experiments1,15,16 with cross-relaxation rates17,18 obtained from molecular dynamics (MD) trajectories; providing formerly inaccessible details.
000-fold enhanced, compared to conventional nuclear magnetic resonance (NMR) spectra. Various methods, from para hydrogenation to dynamic nuclear polarization (DNP) to optical pumping,1 enable the creation of hyperpolarized spin states for a wide array of applications. The use of hyperpolarized water to boost signal intensities in biomolecular NMR spectra has recently received ample attention2–5 as dissolving a target biomolecule in a hyperpolarized buffer enables new approaches at substantially enhanced sensitivity: residue- and time-resolved protein NMR,6 2D and 3D correlation spectra,7 protein folding and folding monitoring,8 membrane interactions,9 structural dynamics,10 and exchange processes.11 However, the understanding of the hyperpolarization transfer mechanism12 from the buffer to the targets remained incomplete; not least as data acquisition in the, most often used, dissolution DNP (dDNP) experiments is typically limited to short periods of 1–2 min that do not allow for determining the exchange pathways via complex pulse sequences. Important insights into hyperpolarization transfer by nuclear Overhauser effects (NOE) between water protons and molecules dissolved therein have, e.g., been achieved by Marco-Rius et al. (13C to 1H)13 or Hu et al. (1H to 1H or 19F).14 To this understanding, we here add a description of the interplay between direct solvent-solute NOE and exchange-relayed NOE in hyperpolarized water; an interesting phenomenon which invokes shifted weights of the different contributions to the magnetization flow as NOE and chemical exchange are differently affected by spin-hyperpolarization. Herein, we provide an understanding of these effects and, hence, also of the transient signal enhancements in biomolecular dDNP. In a dDNP experiment, the sample is ex situ pretreated to boost the spin resonances before transfer to an NMR spectrometer for detection. As the resulting spin-hyperpolarization after the transfer constitutes a non-equilibrium state, its lifetime is necessarily limited. We here demonstrate how to unravel the active transfer mechanisms despite a short time window by integrating dDNP experiments1,15,16 with cross-relaxation rates17,18 obtained from molecular dynamics (MD) trajectories; providing formerly inaccessible details.
Experimentally, we employed the dDNP protocol detailed in ref. 19 (see the ESI†). In brief, we hyperpolarized a water-glycerol mixture20 using a prototype system21 operating at a temperature of TDNP = 1.4 K and a magnetic field of B0,DNP = 6.7 T. After build-up of the 1H spin hyperpolarization, the sample was dissolved with a burst of D2O and transferred to a conventional 11.7 T NMR spectrometer for detection at 298 K. The hyperpolarized sample was then mixed in the NMR tube waiting in the spectrometer with a solution of the target molecules under study. After mixing, the hyperpolarization is readily transferred from the water to the target. As a model target to study the transfer mechanisms, we chose the ubiquitous amino acid arginine due to its widespread use and its variety of labile as well as non-exchanging protons.
Fig. 1 outlines the potential polarization transfer pathways: chemical proton exchange, direct water-to-target NOE, and exchange-relayed NOE. Phenomenologically, the magnetization transfer upon dissolution in hyperpolarized water results in a transient change of the arginine 1H NMR intensities. Fig. 2a and b show the resulting data. Detection started 2 s after mixing to allow the solution to settle before acquisition. The resonances of H2O, and Arg-Hβ, Hγ, as well as Hδ can be discerned. However, the strong water resonance covered Hα. As hyperpolarization is a non-equilibrium state, the water signal decays exponentially to its thermal equilibrium value within ca. 40 s (Fig. 2c). During this period, the non-equilibrium polarization is transferred to the target. We observed that for t < 5 s, all arginine signal intensities are reduced compared to thermal equilibrium Fig. 2b. Then, the signals rise over their equilibrium values during a time of ca. 40 s before reaching the thermal equilibrium. Fig. 2d and e show the negative signal enhancement computed as 1/ε where ε corresponds to the ratio of the signal amplitude S(t) at time t and the thermal equilibrium amplitude STE. All observed arginine protons, Arg-Hβ, Hγ, and Hδ do not chemically exchange with the solvent and yet show substantial signal amplitude changes during this period. Note that the water signal decay is strictly monotone, which outrules biases in signal intensities due to incomplete mixing or convection. The ESI,† contain similar data for poly-aspartate, as a proof-of-concept that the reported method can be applied to other amino acids and macromolecular systems, too.
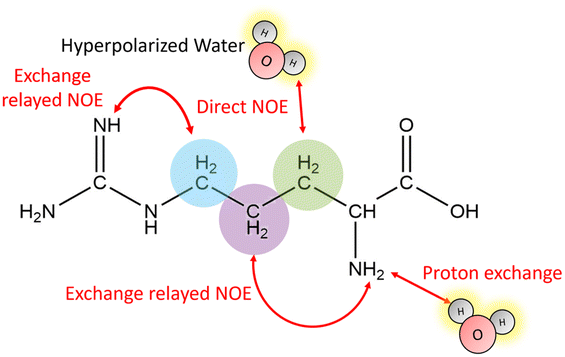 | ||
| Fig. 1 Hyperpolarization transfer pathways between water and arginine: direct nuclear NOE between the solvent and the target, chemical proton exchange of labile moieties, or exchange-relayed NOE. The β-,γ and δ-hydrogens are indicated by the green, violet and blue spheres, respectively. | ||
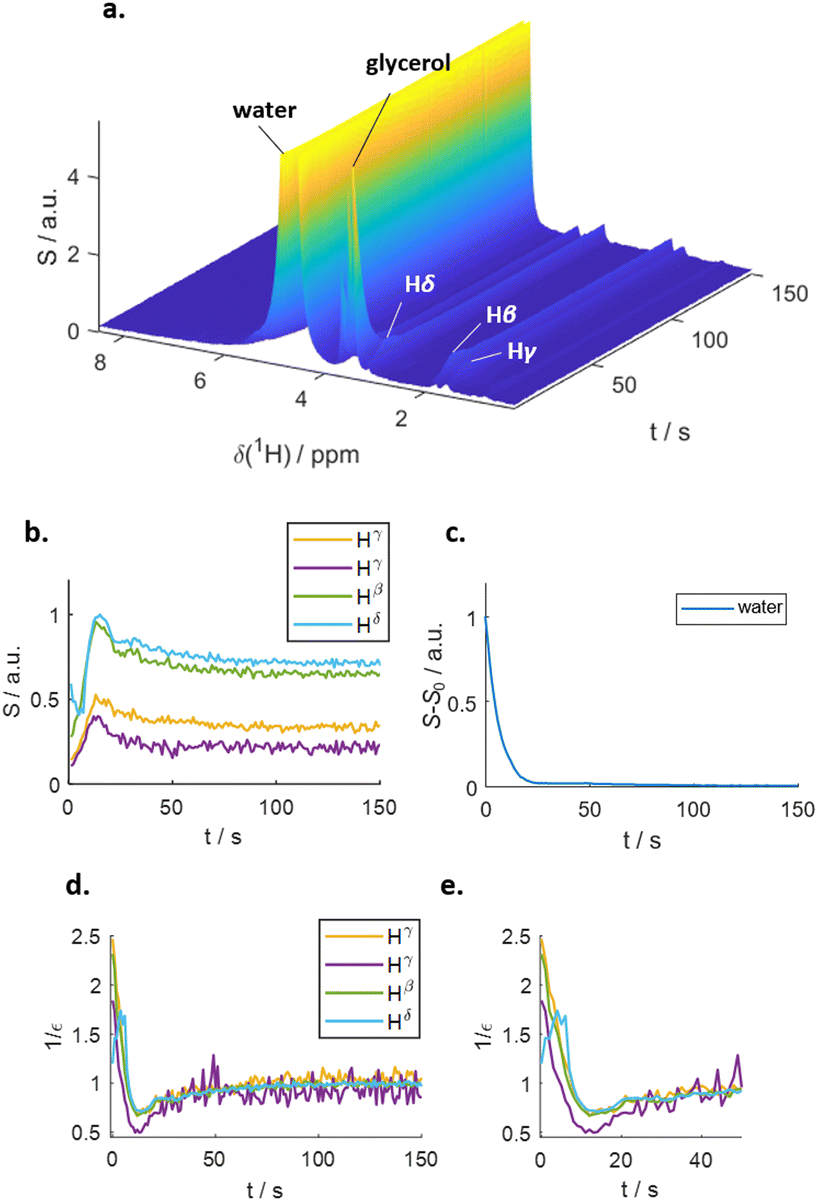 | ||
| Fig. 2 (a) Time series of proton NMR spectra of arginine in hyperpolarized water. t = 0 corresponds to the start of the acquisition period. Next to the aliphatic protons, the water and glycerol lines are visible. (b) Intensities of the aliphatic arginine protons. An initial negative enhancement for 5 s can be discerned, prior to a positive enhancement at t > 5 s. (c) Intensity of the water signal. The decay is strictly monoexponential. (d) Negative signal enhancements 1/ε vs. time. (e) The first 50 s of panel d. | ||
The observed behavior correlates with the well-documented22,23 superposition of a negative direct NOE between the solvent and the target that dominates at the beginning of the time trace and reduces the arginine signal amplitudes and the effects of chemical exchange and exchange-relayed NOE that dominates during the later stages of the trajectory and increases the signal amplitudes. The initial negative enhancements εi are listed in Table 1.
| Atom | 1/εi [a.u.] | σ L [a.u.] | Distance < 6 Å | |
|---|---|---|---|---|
| # H2O | τ [ps] | |||
| Hα | — | 4.98 | 24 | 8.3 |
| Hβ | 2.30 | 4.95 | 24 | 7.6 |
| Hγ | 2.22 | 4.70 | 23 | 7.2 |
| Hδ | 1.24 | 4.55 | 23 | 6.5 |
The differences in build-up and decay rates in Fig. 2b–e result from proton position-dependent efficiencies of hyperpolarization transfer, either via direct or exchange-relayed NOE. To corroborate this interpretation as well as that of the negative signal enhancement at t < 5 s as direct NOE between solvent and target, we employed MD simulations (see ESI† for details) to calculate the direct water-arginine polarization exchange using Redfield relaxation theory.24
The first 20 ns of each trajectory were discarded to avoid the inclusion of possible slow equilibration artifacts. To compute cross-relaxation rates, five hundred starting points were evenly spaced along the trajectory for each correlation function. At each starting point, the distance of the spin pairs was minimized by centering the reference amino acid spin in the simulation box. Subsequently, the coordinates of all molecules were unfolded to undo the coordinate jumps caused by periodic boundary conditions, restoring the natural diffusive motion of the spin pairs. The resulting correlation functions were Fourier transformed using NumPy, and the cross-relaxation rates were averaged over all arginines.
For the computation of the NOE, we capitalized on the fact that the magnetization transfer between two interacting spins I and S with a nuclear spin of 1/2 (e.g. an arginine hydrogen atom and a water hydrogen atom) takes place strictly via a dipole-coupled mechanism. The corresponding time correlation function
 | (1) |
![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) IS joining the two interacting spins I and S; its randomization rate is determined by the translational diffusion of these two spins towards or away from each other. Typically, this is the numerically dominant term in intermolecular NOEs.25 The second term depends on the angle θIS swept by the spin-joining vector during time t. This term represents the gyration of the two spins around a common center. The spectral density JIS(ν) can be obtained by real-part Fourier transformation
IS joining the two interacting spins I and S; its randomization rate is determined by the translational diffusion of these two spins towards or away from each other. Typically, this is the numerically dominant term in intermolecular NOEs.25 The second term depends on the angle θIS swept by the spin-joining vector during time t. This term represents the gyration of the two spins around a common center. The spectral density JIS(ν) can be obtained by real-part Fourier transformation | (2) |
| σNOESYL(ν) = 0.6JIS(νI + νS) − 0.1JIS(|νI − νS|) | (3) |
The spectral density function in eqn (2) is a function of the distance of the interacting species. The distance therefore also affects σNOESYL.28 The most important contributions to σNOESYL should be covered within a distance of 6 Å.28 Using this limit, we counted approximately 24 water molecules in the vicinity of the Hα, Hβ, Hγ and Hδ protons. The corresponding residence times (see Table 1) correlate well with the NOE relaxation rate as shown in the radio plot in Fig. 3.
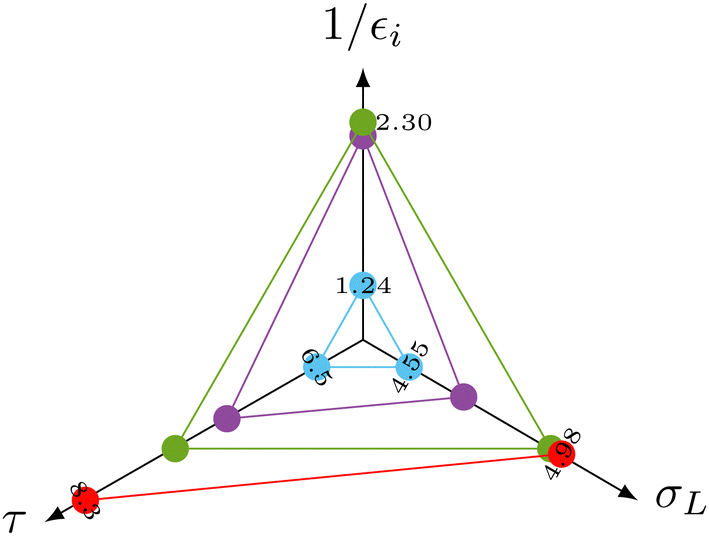 | ||
| Fig. 3 Radio plot of the correlation between the experimental negative signal enhancement immediately after start of the detection 1/εi, the NOE relaxation rate σL and the residence time τ of the interaction water molecules. Hα (red) was masked in the experiments by the broadened water line. | ||
More importantly, the measured initial negative signal enhancements also correlate with the computed values (Fig. 3) corroborating our interpretation of the signal reduction in the dDNP experiments as direct solvent-to-target NOE. The Hβ (green dots) atoms showing the strongest initial negative enhancement 1/εi lead to the highest rates σL. In contrast, water molecules near the arginine Hδ (blue dots) led to a much reduced 1/εi as accompanied by slower NOE transfer and shorter residence times. Hγ (purple dots) consistently laid between Hβ and Hδ for all three parameters.
However, the correlation of computed and experimental data suffers from the drawback that chemical exchange is neglected in our simulations. Hence, we further complemented our conclusions with conventional NOESY (Nuclear Overhauser Enhancement SpectroscopY). We found that both, direct NOE between the water and the different arginine protons as well as exchange processes, are effectively transferring nuclear spin polarization (Fig. 4; see the ESI† for other mixing times). For all mixing times, negative cross-peaks (relative to negative diagonal peaks) between the labile HN and Hη resonances of arginine and the water indicate chemical proton exchange. In addition, we observed cross-peaks between the water and Hβ, Hγ, as well as Hδ indicating direct NOE between arginine and solvent. Intramolecular NOE between all involved arginine protons could be observed, as expected (Fig. 4).
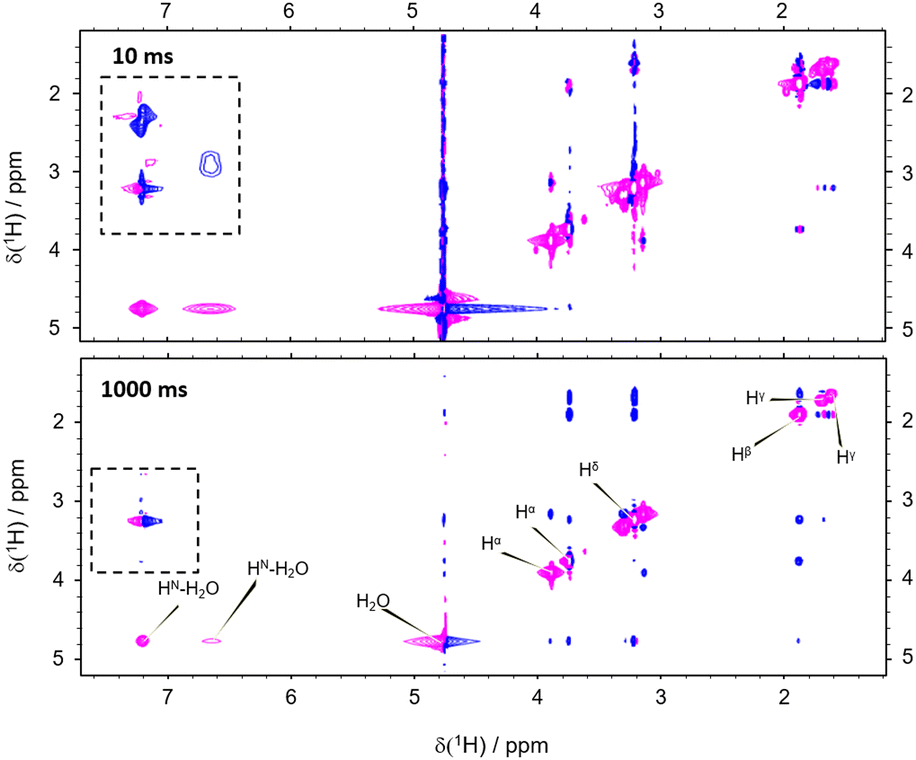 | ||
| Fig. 4 Thermal equilibrium NOESY with a mixing time of 10 ms (top) and 1 s (bottom). Positive cross-peaks between the water and the non-exchangeable protons evidence intermolecular NOE. Cross-peaks between the water and the labile protons indicate proton exchange. The dashed box highlights magnetization transfer between exchanging and aliphatic protons, i.e., exchange-relayed NOE. | ||
Importantly, the NOESY show the simultaneous action of chemical proton exchange and intramolecular NOE leading to exchange-relayed NOE. This is crucial for understanding the hyperpolarization transfer. Since the solvent NOE features an opposed sign relative to the diagonal peaks, an exchange-relayed NOE is necessarily responsible for the positive signal enhancements of the non-exchangeable protons at t > 5 s. The observation of cross-peaks between the signals of the labile HN protons and Hβ, Hγ, as well as Hδ confirms the flow of magnetization from exchanging to non-exchanging moieties (Fig. 4 dashed rectangle).
Summarizing, all three pathways (direct solvent NOE, chemical exchange, as well as exchange relayed NOE) depicted in Fig. 1 can transfer hyperpolarization from the water to a target molecule to boost NMR spectra of biomolecules in a hyperpolarized buffer. For arginine at B0 = 11.7 T, we find negative signal enhancements for all non-labile protons by direct NOE for t < 5 s after dissolution in hyperpolarized water and positive enhancements for t > 5 s by exchange-relayed NOE. As water is a ubiquitous solvent, the reported mechanisms can be readily extended and exploited for other target molecules. The combination of dDNP and computational approaches can thereby be a powerful asset to enlighten both, structural as well as spin dynamics of hyperpolarized solutions. It should be noted that in experiments that detect vividly exchanging protons, as typical for 1H–15N cross-peaks or 15N-edited 1H signals of peptides, nucleic acids, and proteins, the contribution of the negative, direct solvent NOE is canceled by the much more effective proton exchange pathway reaching enhancements of ε > 100. However, for non-labile protons, this effect cannot be neglected and competes with the exchange-relayed pathway. The importance of exchange-relayed effects is further supported by earlier work6 that indicated that backbone 1H–15N signal enhancements are strongest for residues with protic side chains. This observation can readily be explained with exchange-relayed NOE between the side chain and backbone protons.
All raw data are available under https://doi.org/10.5281/zenodo.7113098.
LME, KC and FK performed the experiments. PH developed the software to analyze the MD trajectories. LME, PH and FJ performed simulations. LME, CS and DK analyzed data and conceived the study.
The authors acknowledge support from the NMR core facility of the Faculty of Chemistry, University Vienna, an ERC StG (No 801936), and 2 FWF grants (no. P-33338 N, J-4537 and I4383N).
Conflicts of interest
There are no conflicts to declare.References
- K. V. Kovtunov, E. V. Pokochueva, O. G. Salnikov, S. F. Cousin, D. Kurzbach, B. Vuichoud, S. Jannin, E. Y. Chekmenev, B. M. Goodson and D. A. Barskiy, et al. , Chem. – Asian J., 2018, 13, 1857–1871 CrossRef CAS.
- K. W. Lipsø, S. Bowen, O. Rybalko and J. H. Ardenkjær-Larsen, J. Magn. Reson., 2017, 274, 65–72 CrossRef PubMed.
- R. Kircher, H. Hasse and K. Münnemann, Anal. Chem., 2021, 93, 8897–8905 CrossRef CAS PubMed.
- H. Kouno, K. Orihashi, K. Nishimura, Y. Kawashima, K. Tateishi, T. Uesaka, N. Kimizuka and N. Yanai, Chem. Commun., 2020, 56, 3717–3720 RSC.
- Y. Kharbanda, M. Urbańczyk, V. V. Zhivonitko, S. Mailhiot, M. I. Kettunen and V. Telkki, Angew. Chem., Int. Ed., 2022, e202203957 CAS.
- M. Negroni and D. Kurzbach, Commun. Chem., 2021, 4, 1–7 CrossRef.
- G. L. Olsen, O. Szekely, B. Mateos, P. Kadeřávek, F. Ferrage, R. Konrat, R. Pierattelli, I. C. Felli, G. Bodenhausen and D. Kurzbach, et al. , J. Biomol. NMR, 2020, 74, 161–171 CrossRef CAS PubMed.
- J. Kim, R. Mandal and C. Hilty, J. Phys. Chem. Lett., 2019, 10, 5463–5467 CrossRef CAS PubMed.
- A. Sadet, C. Stavarache, M. Bacalum, M. Radu, G. Bodenhausen, D. Kurzbach and P. R. Vasos, J. Am. Chem. Soc., 2019, 141, 12448–12452 CrossRef CAS.
- D. Kurzbach, E. Canet, A. G. Flamm, A. Jhajharia, E. M. Weber, R. Konrat and G. Bodenhausen, Angew. Chem., Int. Ed., 2017, 56, 389–392 CrossRef CAS PubMed.
- O. Szekely, G. L. Olsen, M. Novakovic, R. Rosenzweig and L. Frydman, J. Am. Chem. Soc., 2020, 142, 9267–9284 CrossRef CAS PubMed.
- J. Kim, M. Liu and C. Hilty, J. Phys. Chem. B, 2017, 121, 6492–6498 CrossRef CAS.
- I. Marco-Rius, S. E. Bohndiek, M. I. Kettunen, T. J. Larkin, M. Basharat, C. Seeley and K. M. Brindle, Contrast Media Mol. Imaging, 2014, 9, 82–186 Search PubMed.
- J. Hu, J. Kim and C. Hilty, J. Phys. Chem. Lett., 2022, 13, 3819–3823 CrossRef CAS PubMed.
- S. Jannin, J.-N. Dumez, P. Giraudeau and D. Kurzbach, J. Magn. Reson., 2019, 305, 41–50 CrossRef CAS PubMed.
- J. H. Ardenkjær-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef PubMed.
- P. Honegger and O. Steinhauser, Phys. Chem. Chem. Phys., 2020, 22, 212–222 RSC.
- P. Honegger and O. Steinhauser, J. Chem. Phys., 2020, 152, 024120 CrossRef PubMed.
- C. Hilty, D. Kurzbach and L. Frydman, Nat. Protoc., 2022, 17, 1621–1657 CrossRef CAS.
- O. Szekely, G. L. Olsen, I. C. Felli and L. Frydman, Anal. Chem., 2018, 90, 6169–6177 CrossRef CAS PubMed.
- T. Kress, K. Che, L. M. Epasto, F. Kozak, M. Negroni, G. L. Olsen, A. Selimovic and D. Kurzbach, Magn. Reson., 2021, 2, 387–394 CrossRef.
- K. Wüthrich, G. Otting and E. Liepinsh, Faraday Discuss., 1992, 93, 35–45 RSC.
- Y. Wang, J. Kim and C. Hilty, Chem. Sci., 2020, 11, 5935–5943 RSC.
- A. G. Redfield, IBM J. Res. Dev., 1957, 1, 19–31 Search PubMed.
- D. Braun, M. Schmollngruber and O. Steinhauser, J. Phys. Chem. Lett., 2017, 8, 3421 CrossRef CAS PubMed.
- A. Abragam, Principles of nuclear magnetism (International series of monographs on physics), Clarendon Press, Oxford, 1961 Search PubMed.
- R. Kimmich, NMR: tomography, diffusometry, relaxometry, Springer Science & Business Media, 2012 Search PubMed.
- P. Honegger, M. E. Di Pietro, F. Castiglione, C. Vaccarini, A. Quant, O. Steinhauser, C. Schröder and A. Mele, J. Phys. Chem. Lett., 2021, 12, 8658–8663 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc03735a |
| This journal is © The Royal Society of Chemistry 2022 |