
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe taming of Clar's hydrocarbon
Leoš
Valenta
 a and
Michal
Juríček
*ab
a and
Michal
Juríček
*ab
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland. E-mail: michal.juricek@chem.uzh.ch
bPrievidza Chemical Society, M. Hodžu 10/16, 971 01 Prievidza, Slovak Republic
First published on 29th August 2022
Abstract
Triangulene is the smallest non-Kekulé graphene fragment known as Clar's hydrocarbon. Due to its open-shell electronic structure, triangulene is a promising molecular building block of carbon-based organic materials for spintronics and quantum molecular science. It comprises six benzenoid rings arranged in a triangular shape with two unpaired electrons delocalized over the entire conjugated core, making this molecule highly reactive. A triplet ground state is predicted for this hydrocarbon by Ovchinnikov's rule, or Lieb's theorem, in accord with Hund's rule. The pioneering work on triangulene was performed almost 70 years ago by Erich Clar, who attempted to prepare the pristine compound. Since then, several synthetic approaches to prepare this molecule have been exploited. The extreme reactivity of triangulene can be circumvented using on-surface techniques or by installation of sterically demanding substituents, which kinetically stabilize the diradical core against oligomerization in solution. The first two examples of a persistent derivative of triangulene were simultaneously and independently developed last year. This article presents a historical development in the synthesis of triangulene and its derivatives and outlines possible future applications in ferromagnetic materials, electrically conductive polymers or quantum computing.
 Leoš Valenta (left) and Michal Juríček (right) | Leoš Valenta received his Bachelor and Master's degrees (2018, with honors) from the University of Chemistry and Technology Prague (Czech Republic). Currently, he is completing his PhD studies at the University of Zurich (Switzerland) in the group of Michal Juríček. His research interests include the synthesis of open-shell molecules and application of their intriguing properties in the development of new materials. Apart from the lab work, he likes good beer and red pandas. Michal Juríček received his Master's degree (2005) from the Comenius University in Bratislava (Slovakia) and his PhD (2011) from the Radboud University Nijmegen (The Netherlands) mentored by Professor Alan Rowan. After a postdoctoral stay in the group of Professor Sir Fraser Stoddart at Northwestern University (United States), he started his independent research career at the University of Basel (Switzerland) hosted in the group of Professor Marcel Mayor. From 2017, he is an assistant professor at the University of Zurich. His group designs, synthesizes and investigates functional organic molecules based on open-shell nanographenes for applications in spintronics. |
Introduction
Isolation of graphene by mechanical exfoliation of graphite1 and identification of its unique characteristics2–5 triggered research on atomically precise and monodispersed graphene fragments.6,7 These molecular systems represent ideal models to guide our understanding of the relationship between the properties and structural parameters like size, shape and edge topology in graphene-based materials.8–11 Among the variety of graphene nanostructures, open-shell graphene fragments12–15 have recently emerged as promising materials to transform conventional electronics into spintronics.16,17 It is envisioned that the spins of unpaired electrons can function as information carriers, and spin-based devices made of these materials can meet the increasing demand for speed and miniaturization in information processing.18,19 The main advantage of graphene-based materials over metals is carbon's small spin–orbit coupling, a prerequisite for achieving long spin-state lifetimes at room temperature.20,21 The unpaired electrons in open-shell graphene fragments are typically highly delocalized over the entire structure, enabling spin–spin communication, which is in contrast to spin-localized nitroxyl radicals, another class of compounds investigated as spin carriers.22,23Considering the bonding situation, two basic types of graphene fragments are recognized, Kekulé and non-Kekulé.24 The Kekulé systems satisfy the standard rules of valence and they can be represented by at least one Kekulé resonance structure in which all electrons are paired in the form of conjugated double bonds. In contrast, no Kekulé resonance structure with all electrons paired can be drawn for the non-Kekulé systems. They contain two or more unpaired π-electrons and do not conform to the standard rule of valence. The Ovchinnikov's rule,25 or the Lieb's theorem,26 can be used to distinguish between the Kekulé and non-Kekulé graphene fragments (Fig. 1). Applying this rule, a star is placed on every other sp2 carbon atom such that no two stars are adjacent. For Kekulé molecules, the number of starred (Ns) and unstarred (Nu) carbon atoms is equal (Ns = Nu), whereas for non-Kekulé molecules, the number of starred carbon atoms is larger than the number of unstarred atoms (Ns > Nu). The difference Ns − Nu equals to the number of unpaired electrons in the molecule and the stars denote all positions where the unpaired electrons can reside considering all resonance structures. In accord with the Hund's rule, each non-bonding orbital is occupied by one electron, where all unpaired electrons have parallel spins, resulting in the ground state of the highest possible multiplicity.
 | ||
| Fig. 1 The Ovchinnikov's rule applied to non-Kekulé triangulene (left) and its Kekulé isomer anthanthrene (right). N = number of starred (s) and unstarred (u) atoms. | ||
The prototypic textbook example of a non-Kekulé molecule is triangulene (Fig. 1), also known as Clar's hydrocarbon, named in the honor of Erich Clar (Fig. 2), who pioneered the field of polycyclic aromatic hydrocarbons and proposed the structure of diradical triangulene in 1953.27,28 Triangulene's triplet ground state is the result of the presence of two non-disjoint singly occupied molecular orbitals (SOMOs; Fig. 3) and based on multiconfigurational calculations, it is predicted to be 13–16 kcal mol−1 lower in energy than the first singlet excited state.29 The spin density is distributed across the entire core, but mostly around the periphery (Fig. 3). Unlike molecular oxygen, a stable triplet diradical,30 triangulene readily oligomerizes under ambient conditions.27,31 The high reactivity is the reason why Clar's attempts to generate pristine triangulene were unsuccessful and why it took several attempts in the following almost 70 years to “tame” this iconic molecule.
 | ||
| Fig. 2 Erich Clar “looking for polycyclic aromatic hydrocarbons”. The photographs from Dr Winfried Willicks (1926–2010) are a courtesy of Glasgow University, School of Chemistry. | ||
 | ||
| Fig. 3 The non-disjoint singly occupied molecular orbitals (SOMOs; left, center) and the spin-density distribution (right; positive/black, negative/white) in triangulene. The non-disjoint SOMOs have atoms in common, which results in the triplet ground state. | ||
In the past five years, two important milestones in the research on triangulene were reached. In 2017, pristine triangulene was generated, visualized and studied on a single-molecule level using scanning probe microscopy on various surfaces under ultra-high vacuum by Pavliček et al.32 In 2021, two substituted triangulene derivatives persistent in oxygen-free solutions at room temperature were synthesized and characterized independently by Shintani et al. and our research team (Juríček et al.).33,34 Considering the great potential of triangulene and related compounds to be employed as building blocks in quantum spin materials, demonstrated recently by Fasel et al. (2021),35 and the methodology that is now available for the in-solution synthesis of triangulene derivatives, we think it is timely to reflect on the development of triangulene chemistry over the past seven decades, as we finally enter the era of solution-processed bulk materials built from persistent open-shell graphene fragments.
The era of Erich Clar (1950s)
The first strategies to prepare the pristine triangulene diradical were developed in the early 1950s by Erich Clar.27,28 Even though he successfully prepared several direct precursors of triangulene, he was not able to isolate the target compound on account of its high reactivity.The first synthetic route began with the addition of phenyllithium to an acyl chloride yielding a tris(tolyl) methanol (Scheme 1). After a series of oxidation and reduction steps, an anthrone derivative with a lactone moiety was obtained, which was reductively opened with an ethanolic solution of KOH and Zn to give a diacid, and then cyclized in the presence of acid yielding triangulene-4,8-dione, the first triangulene precursor of Clar. The third oxidation step with HNO3 was later described as an extremely explosive reaction with low reproducibility, which is the drawback of this otherwise elegant route. The lactone obtained in this step can also provide 4,8,12-trioxotriangulene (TOT), the second triangulene precursor of Clar, upon treatment with Cu in H2SO4. The advantage of this method is a simple work-up; the acidic solution is poured over ice and the pure product is filtered off. One of the Clar's attempts to prepare triangulene was by dehydrogenation of hexahydro triangulene, which was prepared via a reduction of TOT with a Zn powder in a molten mixture of NaCl and ZnCl2 at 300 °C (Scheme 1).27 Clar reported that during the final dehydrogenation, the dihydro precursor of triangulene was observed by means of UV–Vis spectroscopy, however, only a small amount of unreacted starting material was isolated by the end of the reaction and no other triangulene derivatives were detected. Clar concluded that upon exposing the reaction mixture to air, triangulene was most probably formed, but its isolation was not possible. As he states in his seminal work: “The complete polymerization of triangulene under conditions, in which even the most reactive aromatic hydrocarbons like hexacene or 1,2-benzohexacene can be prepared, indicates that triangulene is an unstable diradical.”27
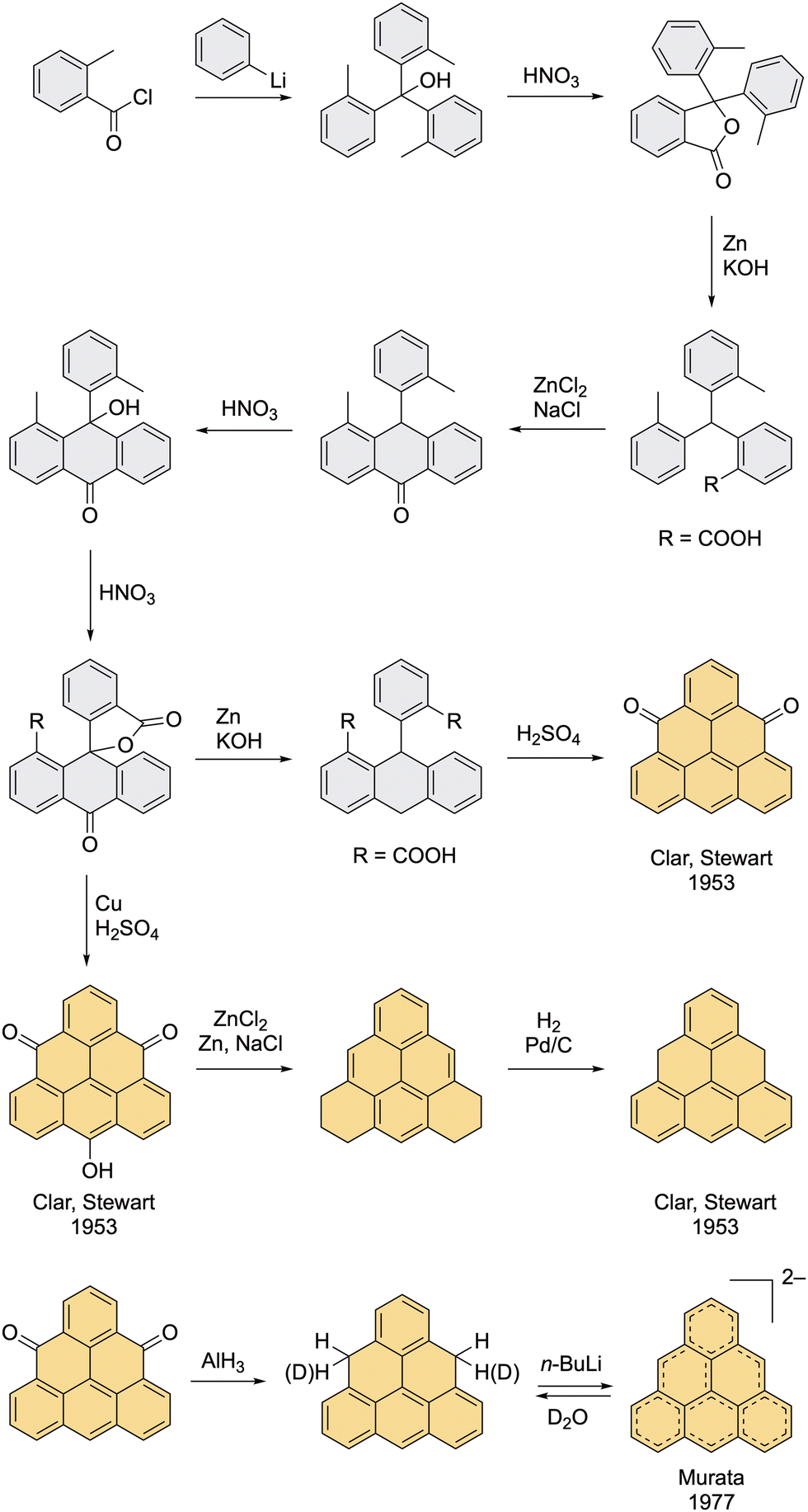 | ||
| Scheme 1 The original synthesis by Clar and Stewart (top) and the synthesis of triangulene dianion by Murata et al. (bottom). | ||
Murata's dianion (1977)
Due to the presence of two degenerate non-bonding orbitals, triangulene can exist not only as a diradical but also as a dication or a dianion. This amphoteric feature was demonstrated by Murata et al. in 1977.36 Clar's precursor triangulene-4,8-dione was reduced with AlH3 to a dihydro precursor of triangulene, which upon careful deprotonation with n-BuLi in degassed d8-THF at −78 °C yielded the desired triangulene dianion (Scheme 1). A highly symmetric 1H NMR spectrum was observed, which supports the D3h symmetry of triangulene dianion. Its formation was also confirmed by HRMS in an experiment, where the sample of dianion was quenched with D2O.Bushby's trianion diradical (1993)
Clar's original synthesis of TOT was improved by Bushby et al., who used TOT as a precursor to prepare the first diradical based on the triangulene scaffold, namely, TOT3− (Scheme 2).37 Using Bushby's route, the lactone intermediate that was prepared by Clar in two steps (Scheme 1) can be accessed in just one step via addition of o-tolylmagnesium bromide to the phthalic anhydride.38 The authors found out that when the crude mixture containing several impurities is treated with hydrazine, which transforms all side products into soluble compounds, only the desired product remains as an insoluble compound and can be filtered off. This finding allowed a large-scale preparation of the lactone intermediate, which then underwent a reductive opening of the lactone ring. The subsequent oxidation of the methyl groups yielded a tricarboxylic acid, which was then cyclized to the final TOT with H2SO4. The treatment of TOT with a hydroxide solution afforded the potassium and tetrabutylammonium salts, which give the target TOT trianion diradical by two-electron chemical reduction with a Na–K alloy.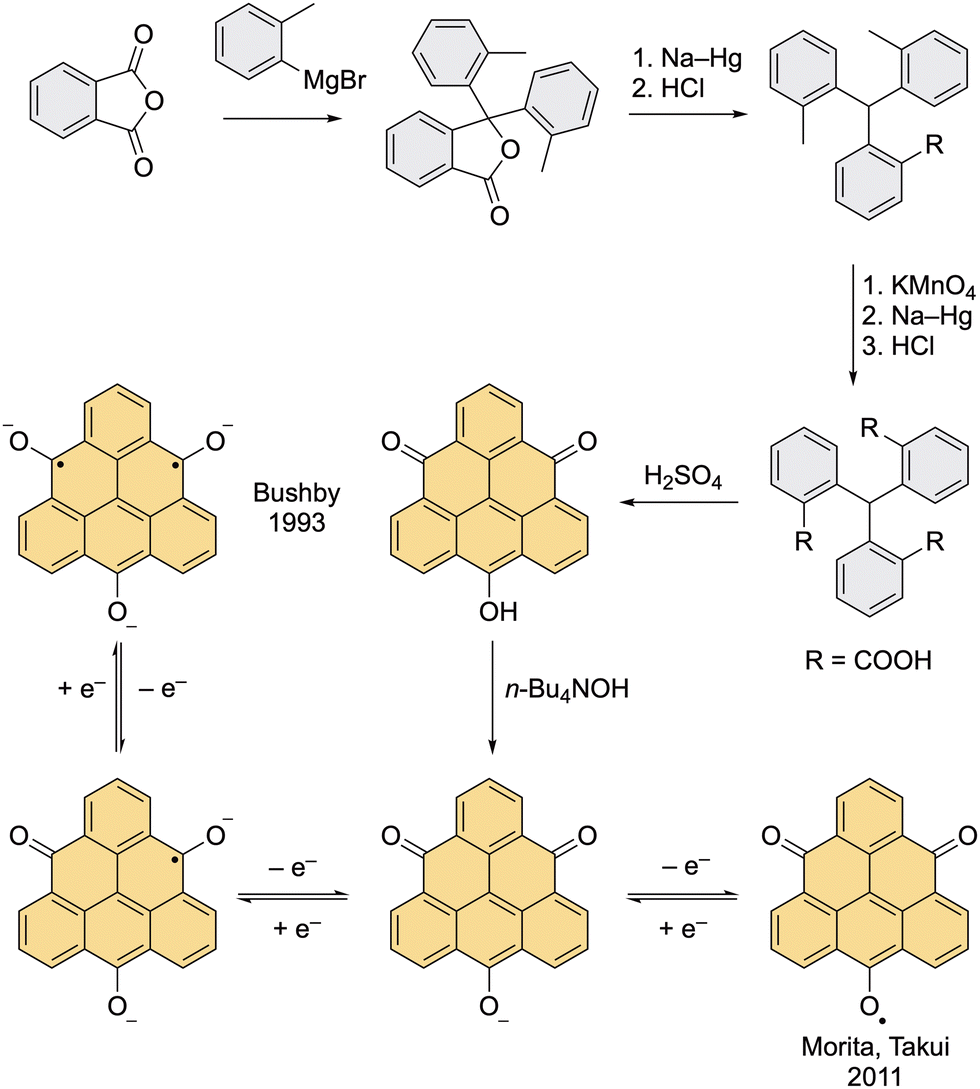 | ||
| Scheme 2 Bushby's improved synthesis of 4,8,12-trioxotriangulene (TOT), and the generation of dianion monoradical and trianion diradical of TOT by Bushby et al. and neutral TOT monoradical by Morita, Takui et al. | ||
Compared with triangulene, the TOT trianion diradical also possesses a triplet ground state but the spin density is additionally delocalized over the oxygen atoms. The triplet ground state was proved by observation of a half-field transition in the EPR spectrum of a frozen DMF solution at 13 K. The trianion diradical is remarkably stable; it can be stored for months in a deoxygenated solution without any loss of intensity of the EPR signal. The increased stability of this diradical compared to triangulene can be explained by the coulombic repulsion between individual diradical molecules, which prevents oligomerization, as well as extended delocalization of the unpaired electrons. Nevertheless, the TOT3− diradical is highly susceptible to the reactions with molecular oxygen.
Morita and Takui's monoradical (2011 – ongoing)
The monoanion of TOT can also undergo a single-electron oxidation with p-chloranil or DDQ to give a neutral monoradical, where the unpaired electron is delocalized over the entire core with the highest SOMO coefficient localized at the central carbon atom (Fig. 4).39 This feature results in an increased stability of this compound and its propensity to form a π-columnar stack, where the molecules are held together by favorable overlap of SOMOs, also known as the pancake bonding (Fig. 4). Over the past 11 years, Morita et al. synthesized various derivatives of this neutral monoradical with different substituents at the vertices of triangulene. It was shown that the tri-tert-butyl derivative could serve as a cathode-active material in coin-type batteries.40 Columnar stacks of TOT monoradical and its derivatives have semiconducting properties with a conductivity of ∼10−3 S cm−1. Mixed salts of the monoradical and the corresponding anion exhibit an even higher conductivity, increased by several orders of magnitude.41 In the most recent work, Morita et al. presented TOT monoradical as a metal-free electrocatalyst for oxygen reduction.42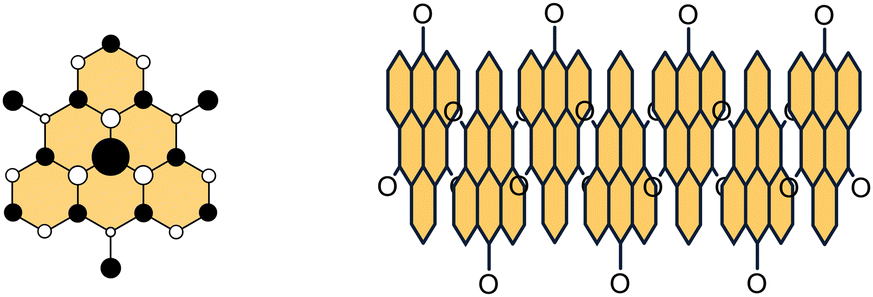 | ||
| Fig. 4 Spin-density distribution in TOT monoradical (left) and columnar stacking of TOT monoradicals (right) via favorable overlap of SOMOs. | ||
Šolomek and Juríček's synthesis (2019)
The synthesis of triangulene-4,8-dione and TOT was further improved by Juríček et al. in 2019 (Scheme 3).43 Compared to Clar's and Bushby's procedures, this method has less steps, employs milder reaction conditions, in particular the explosive oxidation step with HNO3 is avoided, and allows gram-scale preparation of both precursors in an overall yield >50%. The route employs commercially available o-bromobenzyl alcohol that is reacted with n-BuLi before it is quenched with diethyl carbonate. The formed tetraalcohol is oxidized with the Jones reagent to a lactone bearing one aldehyde and one acid moiety, which can be directly cyclized to triangulene-4,8-dione with Cu in H2SO4 or further transformed to a diacid intermediate by means of the Pinnick oxidation and then cyclized with Cu in H2SO4 to TOT. The reduction of triangulene-4,8-dione to the dihydro precursor was performed according to the Murata's procedure with AlH3, however, only low yields (∼10%) were achieved; previously, the yield was not reported.36 For the first time, dihydro triangulene was characterized by means of 2D NMR spectroscopy, which allowed full assignment of the proton and carbon resonances for the two possible isomers formed in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, one with a helicene subunit (major) and one with an anthracene subunit (minor). Notably, dihydro triangulene was additionally characterized in a supramolecular complex with a tetracationic cyclophane ExBox4+ by means of single-crystal X-ray diffraction analysis (Fig. 8, left).
:1 ratio, one with a helicene subunit (major) and one with an anthracene subunit (minor). Notably, dihydro triangulene was additionally characterized in a supramolecular complex with a tetracationic cyclophane ExBox4+ by means of single-crystal X-ray diffraction analysis (Fig. 8, left).
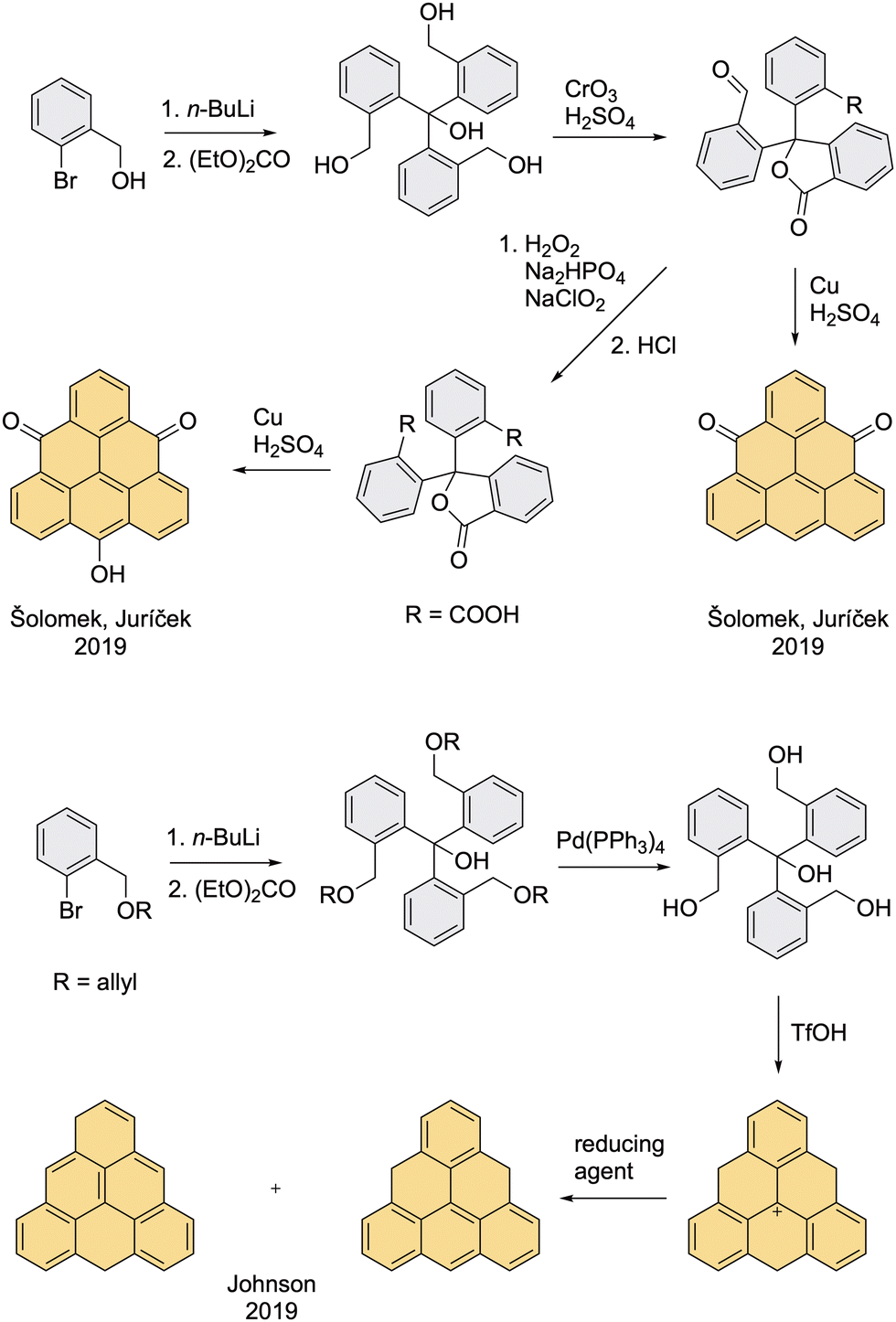 | ||
| Scheme 3 Šolomek and Juríček's improved synthesis of triangulene-4,8-dione and TOT, and Johnson's improved synthesis of dihydro triangulene. | ||
Johnson's synthesis (2019)
In parallel to this work, Johnson et al. reported a different approach for the synthesis of pristine dihydro triangulene, which allows preparation of this triangulene precursor on a gram scale.31 The first step is the same like in the procedure developed by Juríček et al., but the free alcohol groups were protected during the addition with an allyl group, which was removed in the second step. The cyclization of free tetraalcohol with triflic acid provided triangulenyl cation, an unstable intermediate that can be reduced in situ with several different reducing agents, resulting in a mixture of two isomers of dihydro triangulene in a ratio that depends on the reducing agent used (Scheme 4). Combining the procedure of Šolomek and Juríček with that of Johnson, the dihydro precursor of pristine triangulene can now be accessed in two steps with an overall yield of 70% (Scheme 7, top right). | ||
| Scheme 4 Pavliček's synthesis of dihydro triangulene. The AFM image of triangulene is reprinted by permission from Springer Nature: Nature Nanotechnology, Synthesis and characterization of triangulene, N. Pavliček, A. Mistry, Z. Majzik, N. Moll, G Meyer, D. Fox, L. Gross. All rights reserved (2017). | ||
The on-surface era (2017 – ongoing)
A breakthrough in the chemistry of triangulene was the 2017 preparation of pristine triangulene diradical by Pavliček et al.32 Triangulene was generated on an insulating (NaCl and Xe) as well as metallic (Cu) surface by means of atomic manipulation. This methodology combined the in-solution preparation of dihydro triangulene (Scheme 4), which was used for sublimation, and the on-surface generation of the diradical species and their studies. The synthetic strategy was based on the synthesis of Bushby,38 as the 2019 methods (Šolomek, Juríček, Johnson) were not yet available. The main difference compared to Bushby's procedure was that the lactone formed in the first step was not opened reductively but oxidized directly to the diacid lactone with KMnO4 to avoid the use of toxic sodium amalgam. Final reduction with AlH3 using the method developed by Murata et al.36 gave the dihydro precursor, which was sublimed onto a substrate. The very final step, oxidation to triangulene diradical, was performed by means of scanning tunneling microscopy (STM) with a tip positioned above the molecule and application of voltage for several seconds. In many cases, this procedure resulted in a lateral displacement of the molecule, however, in some cases it was possible to obtain atomic force microscopy (AFM) images of triangulene (Scheme 4, bottom left). Although authors performed scanning tunneling spectroscopy (STS) and visualization of the frontier molecular orbitals, it was not possible to determine the electronic ground state of triangulene. This landmark work marks the beginning of the on-surface era in the field of open-shell nanographenes.Since 2017, three π-extended triangulenes (Fig. 5) have been prepared by combining in-solution and on-surface synthesis. In the triangulene series, the number of unpaired electrons (N) can be determined using equation N = n − 1, where n is the number of edge benzenoid rings. Similarly to triangulene, the ground state of the highest possible spin multiplicity is predicted by the Ovchinnikov's rule for this family of nanographenes.
The first π-extended triangulene, [4]triangulene (N = 3), was prepared in 2019 (Fig. 5, left).44 The spin-polarized DFT calculations show three quasi-degenerate SOMOs with a parallel spin alignment of the three unpaired electrons, giving rise to an open-shell quartet ground state. Comparison of the experimental high-resolution STM and STS data with the DFT calculations suggests that [4]triangulene does not interact with the Au(111) surface and maintains the expected quartet ground state when adsorbed on Au(111). Similar behavior was observed for [5]triangulene (N = 4; Fig. 5, center), where STS confirmed the open-shell character of this molecule containing four unpaired electrons.45 Contrary to these results, the latest homolog [7]triangulene (N = 6) possesses a closed-shell electronic structure on Cu(111) (Fig. 5, right), due to the interaction of the molecule with the surface.46 The interaction was probed by high-resolution STM imaging, which revealed a non-planar geometry on the surface. In addition, STS measurements in conjunction with DFT calculations unveiled charge transfer between Cu(111) and [7]triangulene with a considerable hybridization of the molecular orbitals.
 | ||
| Fig. 5 Family of extended triangulenes prepared by means of on-surface synthesis; (from left to right): [4]-, [5]- and [7]triangulene. | ||
Several other systems based on triangulene were prepared by means of on-surface chemistry. The state-of-the-art method employs 9-(2′,6′-dimethylphenyl)anthracene that is available by means of in-solution synthesis as a structural unit, which is transformed on surface into a triangulene unit by a double oxidative ring closure induced by heat (Scheme 7, top left). Using this methodology, another iconic diradical molecule proposed by Erich Clar, namely, Clar's goblet (Fig. 6, top left), was synthesized by Fasel et al.20 The two unpaired electrons in Clar's goblet, each located in a different half of the molecule, are coupled antiferromagnetically in the ground state, but it is possible to excite the molecule into the ferromagnetic state by applying a certain amount of energy, the so-called exchange coupling energy. This energy was found to be 23 meV, which is higher than the heat dissipation energy given by the Landauer limit (∼18 meV at 300 K).47 This characteristic is a prerequisite for making spin-based logic operations, which makes Clar's goblet a promising building block for spintronic devices.
 | ||
| Fig. 6 Structures of Clar's goblet (top left), triangulene nanostar (top right) and triangulene nanochain (bottom). | ||
The same synthetic approach enables the preparation of triangulene oligomeric structures, namely, macrocycles and chains, which are useful models to explore more complex magnetic phenomena. In 2021, Peña et al. reported a nanostar (Fig. 6, top right) comprised of six triangulene units arranged into a ring, which was found to exhibit a collective spin state generated by the interaction of its 12 unpaired π-electrons, two per unit.48 This system displays antiferromagnetic ordering of the six S = 1 sites, where inelastic electron tunneling spectroscopy revealed three collective spin excitations from the many-body singlet ground state to the triplet excited states.
On the other hand, one-dimensional triangulene chains reported by Fasel et al. in 2021 (Fig. 6, bottom) were employed to validate the concept of the so-called spin fractionalization.35 In the 1980s and 1990s, it was predicted that the terminal units of one-dimensional materials composed of S = 1 spin units would display an S = 1/2 state, even though there is no building block with such a quantum spin number.49–51 This principle was confirmed by demonstrating that the terminal triangulene units exhibited Kondo resonances, which are characteristic spectroscopic fingerprints of objects with S = 1/2 that are in contact with a metal surface.
The on-surface techniques to study triangulene-based systems are important for advancing our understanding of carbon-based magnetism of non-Kekulé molecules. Even though these experiments are performed under very delicate conditions like ultra-high vacuum and low temperatures, these studies bring key fundamental insights. To achieve applicability in terms of bulk material production, there is a need for methods that would bring the chemistry of open-shell nanographenes from the surface to solution. Kinetic stabilization represents an ideal tool to increase the lifetime of these highly reactive species in solution without significantly perturbing the electronic structure. In addition, persistent derivatives of open-shell nanographenes can be employed as building blocks of solution-processed and solid materials, which would enable exploitation of the full potential of these unique molecules.
Morita, Takui and Nakasuji's approach (2001)
The first attempt to kinetically stabilize neutral triangulene by use of sterically demanding substituents was made by Nakasuji et al. in 2001, who installed three tert-butyl groups at the vertices (Scheme 5).52 The dihydro precursor of this trisubstituted triangulene was prepared in nine steps from a pyrene derivative as the starting material. Triangulene diradical was generated by the addition of 10 equivalents of p-chloranil as an oxidizing agent at 195 K. The EPR characterization was performed in a frozen-toluene matrix at 123 K. Even though a fully characterized and resolved EPR spectrum could not be obtained because of overlap with the signal of monoradical species, the D value estimated from the outer wings of the cw EPR spectrum is plausible for the triplet state of triangulene. Upon warming of the EPR sample, a signal belonging to the triplet species disappeared and a new signal from pure doublet species was observed. This implies formation of a triangulene polymer with monoradical terminal units, most likely via positions in the centers of the edges that display the highest spin density, as shown by the calculated spin-distribution map (Fig. 3, right). Since the tert-butyl groups were installed at the vertices of triangulene, they protect only the neighboring positions with a minor distribution of spin density, while the most reactive positions remain exposed. Because the choice of positions for installation of the substituents does not reflect the spin distribution, this derivative exhibits low kinetic stability. This problem can be solved by a better positioning of bulky protecting groups, ideally in the centers of the edges to directly shield the most reactive positions as well as the neighboring positions. Synthetically, such task is not a trivial matter, therefore, a derivative protected with substituents in the centers of the edges has remained unknown for almost 70 years since the pioneering work of Erich Clar.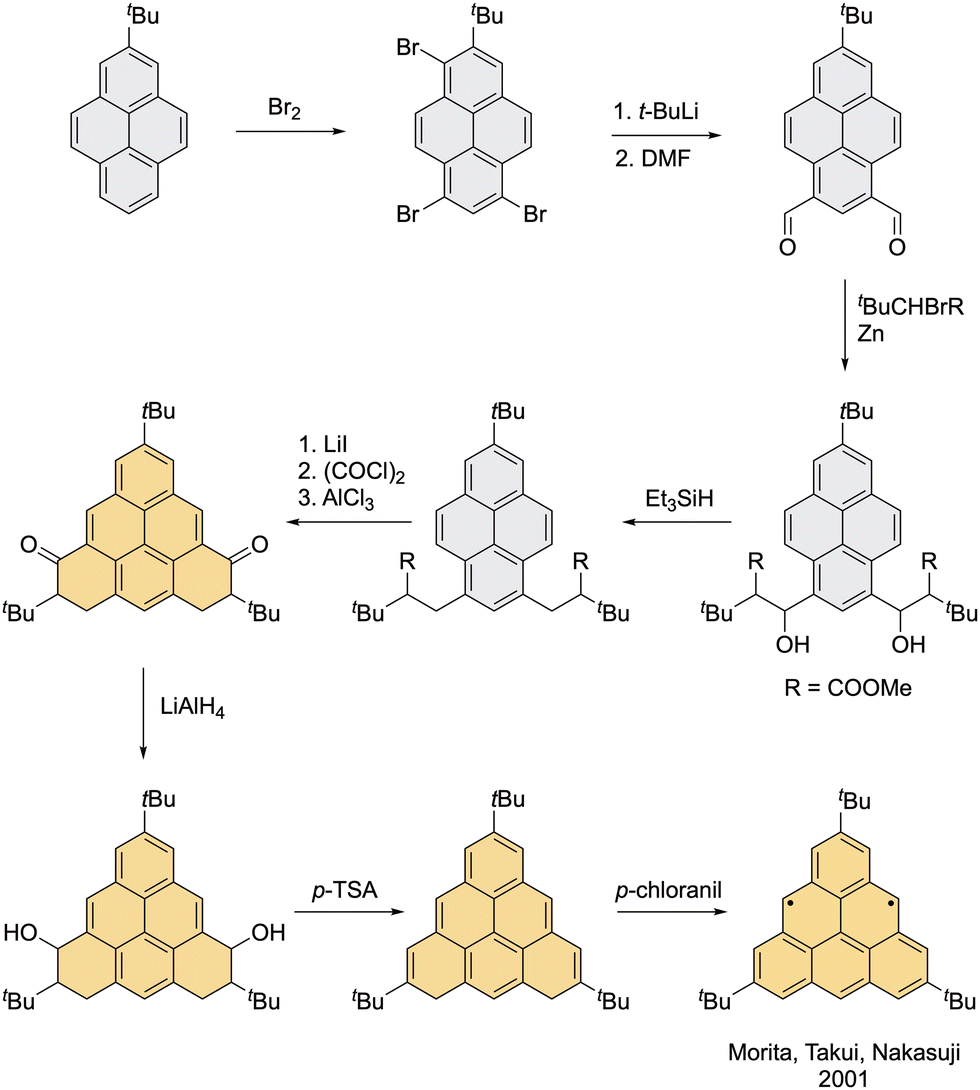 | ||
| Scheme 5 Synthesis of a trisubstituted triangulene derivative equipped with a tert-butyl group at each of the three vertices. | ||
Šolomek and Juríček's attempt (2021)
The first attempt towards this goal was reported by Juríček et al. in 2021.53 In this approach (Scheme 6), the first substituent in the center of one edge was installed by means of the Suzuki cross-coupling reaction of 3,5-di-tert-butylphenylboronic acid and TOT triflate. The resulting monosubstituted triangulene-4,8-dione displays a significantly improved solubility compared to the unsubstituted dione, which is practically insoluble. In the next step, the authors expected that a double 1,2-addition of 3,5-di-tert-butylphenylmagnesium bromide to the carbonyl groups would afford a trisubstituted product, an ideal precursor of a persistent triangulene, where all reactive positions are protected by three substituents in the centers of the edges. Unexpectedly, the product of 1,4-addition was observed when one equivalent of a Grignard reagent was used. The same selectivity was observed also for the second addition. Surprisingly, even the third addition proceeded with a 1,4-selectivity, where instead of the sterically more accessible carbonyl carbon atom (1,2-addition), the nucleophilic attack preferentially takes place at the carbon atom that already bears one bulky aryl substituent, resulting in the formation of an sp3 center. An extensive study of this unexpected reactivity revealed that in the case of carbonyl precursors of non-Kekulé hydrocarbons such as triangulene, there is a strong preference for conjugated additions. This selectivity is dictated by the lowest unoccupied molecular orbital (LUMO), where the coefficient at the 1,4-position is significantly higher than the coefficient at the carbonyl carbon atom. The desired 1,2-addition could not be achieved even upon the addition of an activating agent, for instance, CeCl3. These results suggest that this methodology might not be suitable for the preparation of a derivative bearing substituents in the centers of the triangulene edges.Persistent triangulene derivatives (2021)
Most recently, two teams developed two different synthetic routes to achieve the desired substitution pattern. Juríček et al. presented a triangulene derivative with three mesityl groups at the desired positions,34 whereas Shintani et al. employed three additional tert-butyl groups at the vertices (Fig. 7).33 In both cases, persistency of the triangulene derivative was achieved, as proven by observing no changes in the EPR signal over several weeks in the case of the trisubstituted derivative and, remarkably, by isolating the hexasubstituted derivative and obtaining its solid-state structure by means of single-crystal X-ray crystallography (Fig. 8, right). These results show that while three bulky substituents are sufficient for the stabilization of the triangulene core, additional substituents are beneficial for achieving crystallinity of the triangulene diradical since the solid-state structure of trisubstituted triangulene could not be obtained. | ||
| Fig. 7 Structures of trisubstituted (left) and hexasubstituted (right) triangulene. | ||
Šolomek, Richert and Juríček's derivative
The synthetic approach of Juríček et al. is based on the methodology developed by Wu et al.,45 where the first substituent is introduced in the first step of the synthetic route through the addition of mesitylmagnesium bromide to anthrone (Scheme 7, bottom left). Mesityl was selected as a protecting group because a perpendicular orientation with respect to the triangulene core was expected, minimizing perturbation of the electronic structure by the substituents. The triangulene core was built up in an acid-catalyzed closure of a dialcohol intermediate obtained in seven steps, yielding the dihydro precursor of triangulene. In principle, any nucleophile can be used in the second to last step, where two of the three substituents are installed, which makes this synthetic strategy highly modular. The authors also investigated the possibility of the Suzuki cross-coupling of (10-mesitylanthracen-9-yl)boronic acid, which can easily be prepared in one step from 9-bromo-10-mesitylanthracene, and commercially available 2-bromoisophthalaldehyde. This would make the synthetic pathway significantly shorter, but the desired product could never be observed. The generation of the triangulene diradical was performed with p-chloranil as an oxidizing agent. This transformation is clean, which was shown by the disappearance of signals in the NMR spectrum without formation of any NMR-active side products and by the appearance of exclusively the signal of the diradical in the EPR spectrum. Moreover, a precise addition of a substoichiometric amount of the oxidizing agent or letting the sample of the dihydro precursor stand under ambient conditions allows generation of the triangulene monoradical. The solid-state structure of the monoradical was confirmed by X-ray crystallography (Fig. 8, middle). | ||
| Scheme 6 Nucleophilic additions to triangulene dione. | ||
 | ||
| Fig. 8 Solid-state structures of dihydro triangulene in a supramolecular complex (left), trisubstituted triangulene monoradical (middle) and hexasubstituted triangulene diradical (right). | ||
Shimizu, Shiomi, Sato and Shintani's derivative
Shintani et al. used a different approach, where the triangulene core is built first and the bulky substituents are introduced later on (Scheme 7, bottom right).33 As a starting material, the authors employed TOT with three tert-butyl groups installed at the vertices, which can be prepared in six steps according to the report by Morita et al.40 The details of the experimental procedures that are required to reproduce the synthesis were, however, not described. For this reason, an alternative seven-step synthetic route towards this TOT derivative was developed by Juríček et al. (Scheme 7, bottom right).54,55Interestingly, the first step in the synthesis of hexasubstituted triangulene derivative from trisubstituted TOT is a double nucleophilic addition of mesityllithium in the presence of LaCl3·LiCl as an activating agent promoting 1,2-addition. The reaction without this reagent yielded an undesired product of 1,4-addition. Although in their work Juríček et al. used triangulene-4,8-dione and not TOT, it was predicted that 1,2-selectivity is unlikely to be observed with non-Kekulé molecules. This finding might therefore initiate revisiting of the earlier work and promote studies of the effect of hydroxyl/methoxy groups and bulky tert-butyl substituents in close proximity to the reactive centers on the triangulene dione chemistry. Upon installation of the last substituent, the hexasubstituted triangulene diradical was generated by reductive dehydroxylation of the final dihydroxy precursor with SnCl2 and trifluoroacetic anhydride. The extraordinary stability of this molecule allowed its purification via column chromatography on alumina under a nitrogen atmosphere with benzene and triethylamine as eluents. Single crystals suitable for X-ray diffraction analysis (Fig. 8, right) were grown by recrystallization of the material obtained after column chromatography from a DCM/hexane mixture in a sealed tube in the glovebox. The absence of π–π interactions between the molecules in the crystal suggests minimal magnetic interaction between individual molecules. Despite the high stability of this compound, decomposition was observed when a solution of the diradical was exposed to air. Although the nature of the decomposition product was not determined, the combination of EPR spectroscopy and HR-MS indicates that this structure could be a monoradical derivative bearing one hydroxyl/ketone moiety.
These two seminal works show that triangulene diradical can be generated under both oxidative and reductive conditions. The difference between the two methods is that the oxidation with p-chloranil can provide also the pure hydrocarbon triangulene monoradical, which is not possible via the stepwise reductive dehydroxylation of Shintani's dihydroxy triangulene derivative. The transformation under the oxidative conditions seems to be quantitative without the formation of any side products, as shown by the disappearance of signals in the NMR spectrum and the formation of only one set of signals in the EPR spectrum. In contrast, the reduction with SnCl2 leads to the formation of about 10% of inseparable monoradical impurity. On the other hand, the higher degree of substitution of the triangulene core was found to be crucial for the solid-state isolation of the target triangulene diradical and obtaining single crystals suitable for X-ray diffraction analysis, as the trisubstituted derivative reported by Juríček et al. could only be isolated upon removal of the solvents as an amorphous solid.
Conclusion and future perspectives
Since the pioneering work of Erich Clar in 1950s to this date, three derivatives of neutral triangulene for in-solution studies have been reported. The different substitution patterns clearly show the importance of correct positioning of the blocking groups. Bulky substituents placed at the vertices of triangulene are not sufficient to protect all reactive positions, and tri-tert-butyltriangulene could only be detected in the frozen matrix at low temperatures. A significantly enhanced kinetic stability was achieved by moving the blocking groups from the vertices to the centers of the edges where most of the spin density is localized. Such trisubstituted derivative was shown to be persistent in a deoxygenated solution for weeks without any observable decomposition. The most persistent triangulene derivative was achieved through a combination of both aforementioned approaches. This derivative with six sterically demanding substituents installed at the vertices as well as the centers of the edges was isolated in the solid state and its structure was confirmed by X-ray crystallography.We believe that these two seminal works will motivate the synthesis of analogous open-shell compounds and exploration of the boundaries of their kinetic stability. The two reported methodologies now allow us to pursue questions like “what is the minimum steric bulk of substituents needed to achieve persistency?” or “what substituents are required to maximize the stability of this highly reactive compound or perhaps even achieve stability on air?”. New derivatives with suitable anchor groups might also enable incorporation of triangulene into 2D/3D polymers or single-molecule devices, capitalizing on the unique magnetic properties of the triangulene core. We anticipate that in the near future these materials can be used in the emerging field of quantum computing, electrically conductive polymers or ferromagnetic materials. Despite the fact that the two reported synthetic routes towards persistent triangulene derivatives are different, both are quite lengthy. Therefore, there is a need for developing shorter and more modular synthetic routes to achieve a large-scale production of these promising materials.
 | ||
| Scheme 7 State of the art: current synthetic methodology towards triangulene-based compounds employed on metallic surfaces (top left), the most efficient synthetic pathway towards dihydro precursor of pristine triangulene in solution, taking advantage of the procedures developed by Juríček and Johnson (top right), synthesis of trisubstituted (bottom left) and hexasubstituted (bottom right) triangulene, featuring the synthesis of tri-tert-butyl derivative of TOT developed by Juríček. | ||
Author contributions
L. V. and M. J. co-wrote the manuscript.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
This project received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant Agreement No. 716139) and the Swiss National Science Foundation (SNSF, M.J./PZ00P2_148043, PP00P2_170534 and PP00P2_198900). We would like to thank Prof. Alan Cooper (University of Glasgow) for providing us with the copies of the original photographs of Erich Clar and Nicole Reimann for taking the photographs of us.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178–1271 CrossRef CAS.
- Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 15002 CrossRef CAS.
- X. Xu, K. Müllen and A. Narita, Bull. Chem. Soc. Jpn., 2020, 93, 490–506 CrossRef CAS.
- K. Nakada, M. Fujita, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 17954–17961 CrossRef CAS.
- T. Enoki, Y. Kobayashi and K. I. Fukui, Int. Rev. Phys. Chem., 2007, 26, 609–645 Search PubMed.
- L. Brey and H. A. Fertig, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195408 CrossRef.
- L. Brey and H. A. Fertig, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 235411 CrossRef.
- Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197–204 CrossRef CAS PubMed.
- W. Zeng and J. Wu, Chem, 2021, 7, 358–386 CAS.
- J. Liu and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 23386–23401 CrossRef CAS PubMed.
- Y. Morita and S. Nishida, in Stable Radicals, ed. R. G. Hicks, John Wiley & Sons, Ltd, Chichester, UK, 2010, pp. 81–145 Search PubMed.
- I. Žutić, J. Fabian and S. Das Sarma, Rev. Mod. Phys., 2004, 76, 323–410 CrossRef.
- Z. Bullard, E. C. Girão, J. R. Owens, W. A. Shelton and V. Meunier, Sci. Rep., 2015, 5, 7634 CrossRef CAS PubMed.
- K. Sato, S. Nakazawa, R. Rahimi, T. Ise, S. Nishida, T. Yoshino, N. Mori, K. Toyota, D. Shiomi, Y. Yakiyama, Y. Morita, M. Kitagawa, K. Nakasuji, M. Nakahara, H. Hara, P. Carl, P. Höfer and T. Takui, J. Mater. Chem., 2009, 19, 3739–3754 RSC.
- C. Chappert, A. Fert and F. N. Van Dau, Nat. Mater., 2007, 6, 813–823 CrossRef CAS PubMed.
- S. Mishra, D. Beyer, K. Eimre, S. Kezilebieke, R. Berger, O. Gröning, C. A. Pignedoli, K. Müllen, P. Liljeroth, P. Ruffieux, X. Feng and R. Fasel, Nat. Nanotechnol., 2020, 15, 22–28 CrossRef CAS PubMed.
- S. Mishra, X. Yao, Q. Chen, K. Eimre, O. Gröning, R. Ortiz, M. Di Giovannantonio, J. C. Sancho-García, J. Fernández-Rossier, C. A. Pignedoli, K. Müllen, P. Ruffieux, A. Narita and R. Fasel, Nat. Chem., 2021, 13, 581–586 CrossRef CAS.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2011, 41, 303–349 RSC.
- Q. Wu, P. Zhao, Y. Su, D. Liu and G. Chen, RSC Adv., 2015, 5, 20699–20703 RSC.
- V. I. Minkin, Pure Appl. Chem., 1999, 71, 1919–1981 CrossRef CAS.
- A. A. Ovchinnikov, Theor. Chim. Acta, 1978, 47, 297–304 CrossRef CAS.
- E. H. Lieb, Phys. Rev. Lett., 1989, 62, 1201–1204 CrossRef PubMed.
- E. Clar and D. G. Stewart, J. Am. Chem. Soc., 1953, 75, 2667–2672 CrossRef CAS.
- E. Clar and D. G. Stewart, J. Am. Chem. Soc., 1954, 76, 3504–3507 CrossRef CAS.
- A. Das, T. Müller, F. Plasser and H. Lischka, J. Phys. Chem. A, 2016, 120, 1625–1636 CrossRef CAS PubMed.
- W. T. Borden, R. Hoffmann, T. Stuyver and B. Chen, J. Am. Chem. Soc., 2017, 139, 9010–9018 CrossRef CAS PubMed.
- C. J. Holt, K. J. Wentworth and R. P. Johnson, Angew. Chem., Int. Ed., 2019, 58, 15793–15796 CrossRef CAS PubMed.
- N. Pavliček, A. Mistry, Z. Majzik, N. Moll, G. Meyer, D. J. Fox and L. Gross, Nat. Nanotechnol., 2017, 12, 308–311 CrossRef PubMed.
- S. Arikawa, A. Shimizu, D. Shiomi, K. Sato and R. Shintani, J. Am. Chem. Soc., 2021, 143, 19599–19605 CrossRef CAS PubMed.
- L. Valenta, M. Mayländer, P. Kappeler, O. Blacque, T. Šolomek, S. Richert and M. Juríček, Chem. Commun., 2022, 58, 3019–3022 RSC . Preprint: ChemRxiv, 2021, 10.33774/chemrxiv-2021-gsrtp.
- S. Mishra, G. Catarina, F. Wu, R. Ortiz, D. Jacob, K. Eimre, J. Ma, C. A. Pignedoli, X. Feng, P. Ruffieux, J. Fernández-Rossier and R. Fasel, Nature, 2021, 598, 287–292 CrossRef CAS PubMed.
- O. Hara, K. Tanaka, K. Yamamoto, T. Nakazawa and I. Murata, Tetrahedron Lett., 1977, 18, 2435–2436 CrossRef.
- G. Allinson, R. J. Bushby, J. L. Paillaud, D. Oduwole and K. Sales, J. Am. Chem. Soc., 1993, 115, 2062–2064 CrossRef CAS.
- G. Allinson, R. J. Bushby, J.-L. Paillaud and M. Thornton-Pett, J. Chem. Soc., Perkin Trans. 1, 1995, 385–390 RSC.
- Y. Morita, T. Murata, A. Ueda, C. Yamada, Y. Kanzaki, D. Shiomi, K. Sato and T. Takui, Bull. Chem. Soc. Jpn., 2018, 91, 922–931 CrossRef CAS.
- Y. Morita, S. Nishida, T. Murata, M. Moriguchi, A. Ueda, M. Satoh, K. Arifuku, K. Sato and T. Takui, Nat. Mater., 2011, 10, 947–951 CrossRef CAS.
- T. Murata, C. Yamada, K. Furukawa and Y. Morita, Commun. Chem., 2018, 1, 47 CrossRef.
- T. Murata, K. Kotsuki, H. Murayama, R. Tsuji and Y. Morita, Commun. Chem., 2019, 2, 46 CrossRef.
- P. Ribar, T. Šolomek and M. Juríček, Org. Lett., 2019, 21, 7124–7128 CrossRef CAS.
- S. Mishra, D. Beyer, K. Eimre, J. Liu, R. Berger, O. Gröning, C. A. Pignedoli, K. Müllen, R. Fasel, X. Feng and P. Ruffieux, J. Am. Chem. Soc., 2019, 141, 10621–10625 CrossRef CAS.
- J. Su, M. Telychko, P. Hu, G. Macam, P. Mutombo, H. Zhang, Y. Bao, F. Cheng, Z.-Q. Huang, Z. Qiu, S. J. R. Tan, H. Lin, P. Jelínek, F.-C. Chuang, J. Wu and J. Lu, Sci. Adv., 2019, 5, eaav7717 CrossRef CAS PubMed.
- S. Mishra, K. Xu, K. Eimre, H. Komber, J. Ma, C. A. Pignedoli, R. Fasel, X. Feng and P. Ruffieux, Nanoscale, 2021, 13, 1624–1628 RSC.
- R. Landauer, IBM J. Res. Dev., 1961, 5, 183–191 Search PubMed.
- J. Hieulle, S. Castro, N. Friedrich, A. Vegliante, F. R. Lara, S. Sanz, D. Rey, M. Corso, T. Frederiksen and J. I. Pascual, Angew. Chem., Int. Ed., 2021, 60, 25224–25229 CrossRef CAS PubMed.
- I. Affleck, T. Kennedy, E. H. Lieb and H. Tasaki, Phys. Rev. Lett., 1987, 59, 799–802 CrossRef CAS.
- T. Kennedy, J. Phys.: Condens. Matter, 1990, 2, 5737–5745 CrossRef.
- S. R. White and D. A. Huse, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 3844–3852 CrossRef CAS PubMed.
- J. Inoue, K. Fukui, T. Kubo, S. Nakazawa, K. Sato, D. Shiomi, Y. Morita, K. Yamamoto, T. Takui and K. Nakasuji, J. Am. Chem. Soc., 2001, 123, 12702–12703 CrossRef CAS PubMed.
- P. Ribar, L. Valenta, T. Šolomek and M. Juríček, Angew. Chem., Int. Ed., 2021, 60, 13521–13528 CrossRef CAS PubMed.
- P. Ribar, T. Šolomek, L. Le Pleux, D. Häussinger, A. Prescimone, M. Neuburger and M. Juríček, Synthesis, 2017, 49, 899–909 CrossRef CAS.
- M. G. Rasmussen, M. F. Jespersen, O. Blacque, K. V. Mikkelsen, M. Juríček and M. B. Nielsen, Energy Sci. Eng., 2022, 10, 1752–1762 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |