
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and reactivity of 1-sulfonylcyclooctatriazoles†
Matthew B.
Williams
 ,
Ruaraidh J.
Wells
and
Alistair
Boyer
*
,
Ruaraidh J.
Wells
and
Alistair
Boyer
*
School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: alistair@boyer-research.com
First published on 11th October 2022
Abstract
Strained eight-membered cyclic alkynes undergo rapid inverse electron demand cycloaddition with sulfonyl azides to give the corresponding 1-sulfonylcyclooctatriazoles in excellent yield. Treatment of these sulfonyltriazoles with a chiral rhodium(II) carboxylate catalyst prompted transannular C–H bond insertion in good yield and with excellent ee, or 1,2-H shift.
1-Sulfonyl-1,2,3-triazoles (1-STs) have emerged as valuable building blocks for organic synthesis.1 The combination of nitrogen-rich heteroaromatic and electron-poor sulfonyl group means these compounds have on-demand access to powerful carbene reactivity. In the presence of a catalyst, typically a rhodium(II) carboxylate, the 1,2,3-triazole (3) undergoes denitrogenation to make a metal–carbene complex (4). The route to metal carbenes from 1-STs offers not only complementary reactivity to established carbene methodology but has allowed novel applications and this strategy has been applied to create value-added products, bioactive compounds and natural products. A small selection of 1-ST denitrogenation methodology includes (Scheme 1): functionalising C–H bonds with high yield and excellent selectivity (5),2 heterocycle synthesis (6, 10)3 and 1,2-H shift (11).4
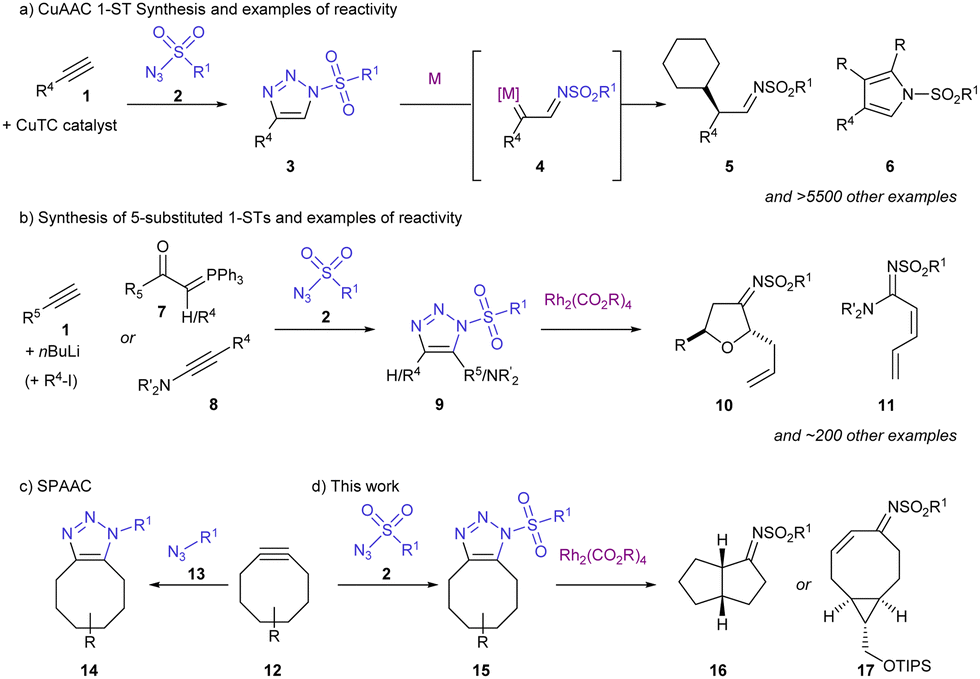 | ||
| Scheme 1 Overview of sulfonyl triazole synthesis and examples of reactivity. | ||
A key consideration in maximising the application of 1-ST methodology is facile access to these valuable heterocycles (Scheme 1). Most commonly, 1-STs with 4-substitution 3 are synthesised by Cu(I) thiophene carboxylate (CuTC) catalysed reaction between an alkyne and sulfonyl azide.5 5-Substituted and 1,4-disubstituted 1-STs 9 have also been synthesised but are less common. These 1-STs 9 can be accessed by anionic methods (using nBuLi),6 Wittig-type reaction (e.g. with 7),7 or others8 but these methods have drawbacks including limited applicability, low yield and poor atom economy. Using a reaction between an electronically matched azide and alkyne pair can also give an efficient route to triazoles. For sulfonyl azides 2, which are very electron-poor, electron-rich alkynes (such as azoalkynes 8) react very rapidly and selectively to form 1-STs.4,9
Increased rate of reaction is observed in the strain-promoted cycloaddition between azides 13 and cyclic alkynes 12 (SPAAC). This “explosive” reactivity was first noted by Blomquist and Liu10 and characterised by Wittig and Krebs.11 Later, Bertozzi realised this cycloaddition would by compatible in vivo and it has become a very popular technique for marking and imaging biological processes.12 However, SPAAC has not been considered in the context of 1-ST synthesis.
Here, the cycloaddition between cyclic alkynes 12 and sulfonyl azides 2 is explored as a route to the less-exploited class of 4,5-disubstituted 1-ST and described computationally. The reactivity of two types of the resulting trisubstituted triazole products 15 has been developed into an enantioselective transannular C–H insertion (16) and 1,2-H shift (17) depending on the cyclic alkyne.
A range of sulfonyl azides 2 was considered in the cycloaddition with cyclooctyne 18 (Fig. 1). Cyclooctyne was readily prepared in three steps from (Z)-cylcooctene13 and it was an important consideration that cyclooctyne decomposes upon storage (over days under argon at −20 °C). An equimolar mixture of cyclooctyne 18 and sulfonyl azide 2 was stirred in dichloromethane and the cycloaddition was complete by 30 min. The cycloadduct 1-ST 20 could be isolated directly from the reaction or following facile purification. For operational convenience, cyclooctyne could also be used in excess to ensure complete conversion and any unreacted excess could be removed in vacuo. The reaction was quantitative to give methanesulfonyl 20a, p-toluenesulfonyl 20b, p-nitrobenzenesulfonyl 20c and p-methoxybenzenesulfonyl 20d triazoles, showing excellent tolerance for across a range of electron-rich to electron-poor sulfonyl azides. The process also gave a very high yield of the triazole with the bulky triisopropylbenzenesulfonyl group 20e as well as polyaromatic sulfonyl triazoles with a p-biphenyl 20f or dimethylaminonaphthyl 20g substituent. Bicyclo[6.1.0]non-4-ynes (BCNs) are a popular class of cyclic alkyne, having a functional handle on the cyclopropyl ring, plus the bicyclic system brings an increase in reactivity through increased strain.14 A silicon protected BCN 19 was prepared in short order. The more functionalised cyclooctyne 19 also gave excellent yields of triazole 21 when reacting with a selection of different alkyl and aryl sulfonyl azides 21a–d.
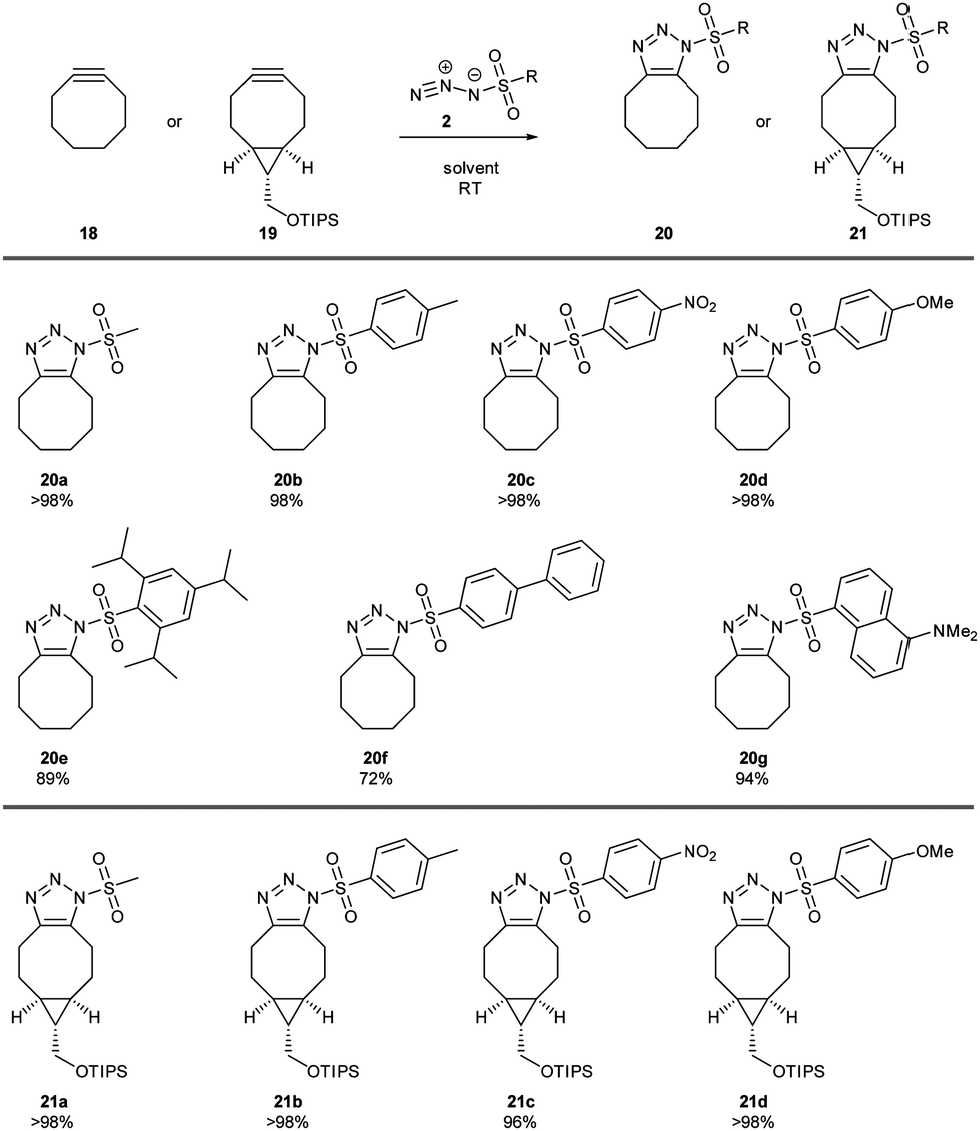 | ||
| Fig. 1 Cycloaddition between MsN32 and cyclooctynes 18, 19. Isolated yield. | ||
The increased reactivity of cyclooctynes is attributed to the alkyne they contain being distorted from its ideal 180° angle. Bickelhaupt and Houk have formalised a distortion–interaction transition state theory for reactions15 that is particularly suited to describe the reactions of strained molecules and has been applied to cyclooctynes.16 In this model, the activation energy required for a reaction between molecules (ΔE‡) is a combination of the energy required for the individual components to distort to their transition state geometries (ΔE‡dist) offset by stabilising orbital interactions between those two components (ΔE‡int = ΔE‡ − ΔE‡dist). The transition states for the cycloaddition between mesyl azide and each of cyclooctyne TS1 BCN TS2 and acetylene TS3 were considered (Table 1) in the framework of Houk's previous work in this area.16a Calculations were performed on the PSI4 program:17 geometry optimisation and thermochemistry analysis was performed using B3LYP/6-31G(d); and orbital energies were calculated using HF/6-311++G(2d,p) at the B3LYP/6-31g(d) geometries.
|
|
||||
|---|---|---|---|---|
| TS1 | TS2 | TS3 | ||
| Calculations at B3LYP/6-31g(d) except orbital energies at HF/6-311++G(2d,p)//B3LYP/6-31g(d). See ESI. | ||||
| ΔE | 8.4 | 7.1 | 16.1 | kcal mol−1 |
| ΔE‡dist (alkyne) | 1.7 | 1.2 | 5.4 | kcal mol−1 |
| ΔE‡dist (azide) | 17.7 | 16.0 | 21.6 | kcal mol−1 |
| ΔE‡int | −11.0 | −10.1 | −10.9 | kcal mol−1 |
| ΔG‡ | 20.6 | 19.2 | 28.2 | kcal mol−1 |
| HOMOalkyne–LUMOazide | 10.5 | 10.6 | 12.1 | eV |
| HOMOazide–LUMOalkyne | 13.0 | 12.9 | 13.0 | eV |

In each of the three transition structures, the cycloaddition was concerted, and asynchronous: with shorter C–N distances at the unsubstituted azide N3 terminus. The azide required significant distortion to reach its reacting geometry: the 175° ∠N–N–N bond angle in MsN3 ground state18 being reduced by over 35° in the three transition structures. This distortion corresponded to a 17.7, 16.0 and 21.6 kcal mol−1 increase in energy (ΔE‡dist) for the azide to attain these reactive geometries. For the cyclic alkynes, only a small deviation from the ground state19 alkyne bond angles was required and the associated change in energy was small (ΔE‡dist = 1.7 and 1.2 kcal mol−1). In contrast, the 180° alkyne bond angles in acetylene were reduced considerably in its transition structure, with an associated ΔE‡dist of 5.4 kcal mol−1. The interaction energy, was very similar across the three reactions (ΔE‡int: −11.0, −8.7; −10.9 kcal mol−1) suggesting that each of the three reactions has similar electrostatic, charge transfer and repulsion interactions. Overall, the  for the cycloaddition of MsN3 with the cyclic alkynes was significantly lower than calculated for acetylene and this analysis suggests that this is due to the strained alkyne being preorganised towards its transition state geometry.
for the cycloaddition of MsN3 with the cyclic alkynes was significantly lower than calculated for acetylene and this analysis suggests that this is due to the strained alkyne being preorganised towards its transition state geometry.
Examination of frontier molecular orbital energies revealed that for each of the transition structures, the HOMOalkyne–LUMOazide gap was lower than the HOMOazide–LUMOalkyne counterpart. This constitutes an inverse electron-demand (IED) mechanism:16c usually for SPAAC the dominant orbital interactions are between the azide HOMO and alkyne LUMO—that can be explained by the very electron withdrawing nature of the sulfonyl group.
As a novel class of 1-ST, the reactivity of the cyclooctatriazoles was probed (Scheme 2). The 1-ST 20b derived from cyclooctyne and TsN3 was treated with rhodium(II) acetate as catalyst in the presence of 2,5-dimethylfuran,3c,i,j styrene,20 triethylsilane,21 or benzonitrile,22 all of which have been demonstrated to be excellent partners for transformations with 4-substituted sulfonyl triazoles. Once the starting material was consumed, THF and lithium aluminium hydride were added to convert any sulfonyl imine into the corresponding amide. In each case the same outcome was observed, a new product was formed that did not incorporate any reacting partner. Analysis by NMR revealed that the product was a [3.3.0]-bicyclic sulfonamide 22b and that it had been formed with complete diastereocontrol (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr by 1H NMR). The eight-membered ring is well known to have close transannular interactions23 so this reaction is proposed to involve 1,5-insertion of the rhodium carbene into the transannular C–H bond.24,25 The same outcome was observed for all the different sulfonyl groups evaluated (22a–g). The stereochemistry was confirmed by comparison with known bicyclic sulfonamide 22b that was accessed by anionic transannular aziridine opening and whose structure was validated by independent synthesis and crystallography.26
:1 dr by 1H NMR). The eight-membered ring is well known to have close transannular interactions23 so this reaction is proposed to involve 1,5-insertion of the rhodium carbene into the transannular C–H bond.24,25 The same outcome was observed for all the different sulfonyl groups evaluated (22a–g). The stereochemistry was confirmed by comparison with known bicyclic sulfonamide 22b that was accessed by anionic transannular aziridine opening and whose structure was validated by independent synthesis and crystallography.26
 | ||
| Scheme 2 Transannular C–H insertion reaction. | ||
The generation of stereocentres prompted an investigation into creating an enantioselective variant of the reaction (Table 2 and ESI†). There are many chiral rhodium(II) carboxylate catalysts that have been demonstrated to impart good enantioselectivity using 1-STs. A brief screen showed that Rh2(S-NTTL)4 catalyst27 was significantly more effective at controlling stereoselectivity in the reaction than Rh2DOSP4 or Rh2PTAD4. Interestingly, the selectivity was also complementary between these ligands. Lowering the temperature from 90 to 50 °C gave better enantioselectivity (29% ee) but at ambient temperature the reaction did not proceed. Varying the solvent gave another step towards enantioselectivity with toluene (68% ee) and perfluorobenzene (91% ee) giving further improvement. Finally, the sulfonyl group was considered. Enantiomeric excess was determined by HPLC analysis following LiAlH4 reduction; or the intermediate sulfonyl imine was hydrolysed to the ketone 23 using basic Al2O3 and converted to the tertiary alcohol 24 by nucleophilic addition of PhMgBr (>20:1 dr by 1H NMR, Scheme 2). Accessing known ketone 2328 allowed further confirmation of the absolute stereochemistry. The second workup was essential in cases where the sulfonamide enantiomers 22 could not be separated by HPLC or functionality was incompatible with LiAlH4 reduction (i.e. Ns, R = pNO2C6H4). Electron donating aromatic sulfonyl groups gave their bicyclic product in lower ee compared with electron withdrawing ones. The Ns group gave higher enantioselectivity and accelerated the reaction rate. Combining these outcomes: treating the N–Ns cyclooctatriazole product with Rh2(S-NTTL)4 in perfluorobenzene at 50 °C followed by the hydrolysis and Grignard addition resulted in clean conversion to the bicyclic product in 94% ee and with 78% yield over the two steps.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Solvent | T | Workup | Yield (%) | ee (%) |
| NTTL = 2-(1,3-dioxobenzo[de]isoquinolin-2-yl)-3,3-dimethyl-butanoate. | ||||||
| 1 | pTol | CH2Cl2 | 50 | LiAlH4 | 36 | 29 |
| 2 | pTol | PhMe | 50 | LiAlH4 | 43 | 68 |
| 3 | pTol | C 6 F 6 | 50 | LiAlH4 | 72 | 91 |
| 4 | pMeOC6H4 | PhMe | 50 | LiAlH4 | 36 | 24 |
| 5 | pNO2C6H4 | PhMe | 50 | Al2O3/PhMgBr | 57 | 69 |
| 6 | pNO2C6H4 | C6F6 | 50 | Al2O3/PhMgBr | 78 | 94 |

The reactivity of the novel N-sulfonylBCNs 21 was also investigated (Scheme 3). In the case of these 1-STs, there was no transannular reaction. Switching to much more forcing conditions was required to see any reaction, namely toluene as solvent, Rh2(OAc)4 as catalyst and 120 °C (sealed vial). At this temperature, the rhodium(II) complex catalysed a denitrogenation and 1,2-H shift to give an α,β-unsaturated intermediate 17; that was hydrolysed to the corresponding ketone 25. The same outcome was observed for a number of different sulfonyl group each of which delivered the enone in good yield. The 1,2-H shift is well-documented in metallocarbenes with an adjacent C–H bond, including those derived from 1-STs.4 It is proposed that the difference in reactivity compared to the cyclooctyne derived 1-STs was that the resulting polycyclic product 26 would be highly strained.
 | ||
| Scheme 3 1,2-H Shift. | ||
In summary, strain-promoted inverse electron demand cycloaddition is an effective route to highly substituted 1-STs. These can undergo transannular C–H insertion with very high enantioselectivity or 1,2-H shift.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) T. Miura and M. Murakami, Rhodium Catal. Org. Synth., 2019, 449–470 CAS; (b) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151–5162 RSC; (c) P. Anbarasan, D. Yadagiri and S. Rajasekar, Synthesis, 2014, 3004–3023 CrossRef CAS.
- (a) S. Chuprakov, J. A. Malik, M. Zibinsky and V. V. Fokin, J. Am. Chem. Soc., 2011, 133, 10352–10355 CrossRef CAS; (b) L. Li, X.-H. Xia, Y. Wang, P. P. Bora and Q. Kang, Adv. Synth. Catal., 2015, 357, 2089–2097 CrossRef CAS; (c) V. N. G. Lindsay, H. M. F. Viart and R. Sarpong, J. Am. Chem. Soc., 2015, 137, 8368–8371 CrossRef CAS; (d) X. Ma, F. Wu, X. Yi, H. Wang and W. Chen, Chem. Commun., 2015, 51, 6862–6865 RSC; (e) M.-H. Shen, Y.-P. Pan, Z.-H. Jia, X.-T. Ren, P. Zhang and H.-D. Xu, Org. Biomol. Chem., 2015, 13, 4851–4854 RSC; (f) M. Senoo, A. Furukawa, T. Hata and H. Urabe, Chem. – Eur. J., 2016, 22, 890–895 CrossRef CAS PubMed; (g) H. Shen, J. Fu, H. Yuan, J. Gong and Z. Yang, J. Org. Chem., 2016, 81, 10180–10192 CrossRef CAS; (h) Z. J. Garlets and H. M. L. Davies, Org. Lett., 2018, 20, 2168–2171 CrossRef CAS PubMed; (i) J. Vaitla, Y. T. Boni and H. M. L. Davies, Angew. Chem., Int. Ed., 2020, 59, 7397–7402 CrossRef CAS; (j) K. Pal and C. M. R. Volla, Chem. Rec., 2021, 21, 4032–4058 CrossRef CAS.
- (a) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862–872 CrossRef CAS; (b) T. Miura, K. Hiraga, T. Biyajima, T. Nakamuro and M. Murakami, Org. Lett., 2013, 15, 3298–3301 CrossRef CAS; (c) B. T. Parr, S. A. Green and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 4716–4718 CrossRef CAS PubMed; (d) E. E. Schultz and R. Sarpong, J. Am. Chem. Soc., 2013, 135, 4696–4699 CrossRef CAS; (e) J. E. Spangler and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 6802–6805 CrossRef CAS; (f) S. W. Kwok, L. Zhang, N. P. Grimster and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53, 3452–3456 CrossRef CAS; (g) T. Miura, Y. Funakoshi and M. Murakami, J. Am. Chem. Soc., 2014, 136, 2272–2275 CrossRef CAS PubMed; (h) H. Shang, Y. Wang, Y. Tian, J. Feng and Y. Tang, Angew. Chem., Int. Ed., 2014, 53, 5662–5666 CrossRef CAS; (i) Y. Funakoshi, T. Miura and M. Murakami, Org. Lett., 2016, 18, 6284–6287 CrossRef CAS PubMed; (j) A. S. Makarov, M. G. Uchuskin and A. S. K. Hashmi, Chem. Sci., 2019, 10, 8583–8588 RSC; (k) M. B. Williams and A. Boyer, J. Org. Chem., 2022 DOI:10.1021/acs.joc.2c00434.
- M. L. Martin and A. Boyer, Eur. J. Org. Chem., 2021, 5857–5861 CrossRef CAS.
- (a) J. Raushel and V. V. Fokin, Org. Lett., 2010, 12, 4952–4955 CrossRef CAS; (b) E. J. Yoo, M. Ahlquist, S. H. Kim, I. Bae, V. V. Fokin, K. B. Sharpless and S. Chang, Angew. Chem., Int. Ed., 2007, 46, 1730–1733 CrossRef CAS PubMed.
- (a) M. E. Meza-Aviña, M. K. Patel and M. P. Croatt, Tetrahedron, 2013, 69, 7840–7846 CrossRef; (b) M. E. Meza-Aviña, M. K. Patel, C. B. Lee, T. J. Dietz and M. P. Croatt, Org. Lett., 2011, 13, 2984–2987 CrossRef; (c) J. Boyer, C. Mack, N. Goebel and J. L. Morgan, J. Org. Chem., 2003, 23, 1051–1053 CrossRef.
- G. R. Harvey, J. Org. Chem., 1958, 31, 1587–1590 CrossRef.
- (a) S. Rajasekar and P. Anbarasan, Chem. – Asian J., 2019, 14, 4563–4567 CrossRef CAS; (b) J. John, J. Thomas and W. Dehaen, Chem. Commun., 2015, 51, 10797–10806 RSC; (c) Y. Y. Morzherin, Y. A. Rozin, E. A. Vorob'eva and V. A. Bakulev, Chem. Heterocycl. Compd., 2001, 37, 560–566 CrossRef CAS.
- (a) R. E. Harmon, F. Stanley, S. K. Gupta and J. Johnson, J. Org. Chem., 1970, 35, 3444–3448 CrossRef CAS; (b) M. Regitz and G. Himbert, Tetrahedron Lett., 1970, 11, 2823–2826 CrossRef; (c) N. Selander and V. V. Fokin, J. Am. Chem. Soc., 2012, 134, 2477–2480 CrossRef CAS.
- A. T. Blomquist and L. H. Liu, J. Am. Chem. Soc., 1953, 75, 2153–2154 CrossRef CAS.
- G. Wittig and A. Krebs, Chem. Ber., 1961, 94, 3260–3275 CrossRef CAS.
- (a) J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS; (b) J. M. Baskin and C. R. Bertozzi, QSAR Comb. Sci., 2007, 26, 1211–1219 CrossRef CAS; (c) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- L. Brandsma and H. D. Verkruijsse, Synthesis, 1978, 290 CrossRef CAS.
- (a) J. G. K. O’Brien, S. R. Chintala and J. M. Fox, J. Org. Chem., 2017, 83, 7500–7503 CrossRef; (b) J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed.
- (a) F. M. Bickelhaupt, J. Comput. Chem., 1999, 20, 114–128 CrossRef CAS; (b) D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 10646–10647 CrossRef CAS PubMed; (c) W.-J. van Zeist and F. M. Bickelhaupt, Org. Biomol. Chem., 2010, 8, 3118–3127 RSC; (d) F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- (a) D. H. Ess, G. O. Jones and K. N. Houk, Org. Lett., 2008, 10, 1633–1636 CrossRef CAS; (b) F. Schoenebeck, D. H. Ess, G. O. Jones and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 8121–8133 CrossRef CAS; (c) J. Dommerholt, O. van Rooijen, A. Borrmann, C. F. Guerra, F. M. Bickelhaupt and F. L. van Delft, Nat. Commun., 2014, 5, 5378 CrossRef CAS.
- J. M. Turney, A. C. Simmonett, R. M. Parrish, E. G. Hohenstein, F. A. Evangelista, J. T. Fermann, B. J. Mintz, L. A. Burns, J. J. Wilke, M. L. Abrams, N. J. Russ, M. L. Leininger, C. L. Janssen, E. T. Seidl, W. D. Allen, H. F. Schaefer, R. A. King, E. F. Valeev, C. D. Sherrill and T. D. Crawford, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 556–565 CAS.
- G. Deng, D. Li, Z. Wu, H. Li, E. Bernhardt and X. Zeng, J. Phys. Chem. A, 2016, 120, 5590–5597 CrossRef CAS.
- I. Yavari, F. Nasiri, H. Djahaniani and A. Jabbari, Int. J. Quantum Chem., 2006, 106, 697–703 CrossRef CAS.
- (a) S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034–18035 CrossRef CAS; (b) T. Miura, T. Nakamuro, Y. Ishihara, Y. Nagata and M. Murakami, Angew. Chem., Int. Ed., 2020, 59, 20475–20479 CrossRef CAS; (c) T. Miura, T. Nakamuro, H. Nikishima and M. Murakami, Chem. Lett., 2016, 45, 1003–1005 CrossRef CAS; (d) Y. Xing, G. Sheng, J. Wang, P. Lu and Y. Wang, Org. Lett., 2014, 16, 1244–1247 CrossRef CAS.
- H. Wang, H. Qiao, H. Zhang, H. Yang, Y. Zhao and H. Fu, Eur. J. Org. Chem., 2015, 4471–4480 CrossRef CAS.
- T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972–14974 CrossRef CAS PubMed.
- (a) A. C. Cope, H.-H. Lee and H. E. Petree, J. Am. Chem. Soc., 1958, 80, 2849–2852 CrossRef CAS; (b) L. A. Paquette, Y. Miyahara and C. W. Doecke, J. Am. Chem. Soc., 1986, 108, 1716–1718 CrossRef CAS.
- (a) G. Engling, T. Emrick, J. Hellmann, E. McElroy, M. Brandt and I. D. Reingold, J. Org. Chem., 1994, 59, 1945 CrossRef CAS; (b) P. Müller and E. Maîtrejean, Collect. Czech. Chem. Commun., 1999, 64, 1807–1826 CrossRef; (c) M. Regitz and J. Rüter, Chem. Ber., 1969, 102, 3877–3890 CrossRef CAS.
- (a) D. M. Hodgson, T. J. Buxton, I. D. Cameron, E. Gras and E. H. M. Kirton, Org. Biomol. Chem., 2003, 1, 4293–4301 RSC; (b) D. M. Hodgson and I. D. Cameron, Org. Lett., 2001, 3, 441–444 CrossRef CAS; (c) P. Müller and P. Nury, Helv. Chim. Acta, 2001, 84, 662–677 CrossRef; (d) D. Stead, P. O'Brien and A. Sanderson, Org. Lett., 2008, 10, 1409–1412 CrossRef CAS.
- (a) P. Müller, D. Riegert and G. Bernardinelli, Helv. Chim. Acta, 2004, 87, 227–239 CrossRef; (b) P. O’Brien, C. M. Rosser and D. Caine, Tetrahedron Lett., 2003, 44, 6613–6615 CrossRef; (c) P. O'Brien, C. M. Rosser and D. Caine, Tetrahedron, 2003, 59, 9779–9791 CrossRef.
- F. G. Adly, J. Maddalena and A. Ghanem, Chirality, 2014, 26, 764–774 CrossRef CAS.
- (a) D. C. Billington, W. J. Kerr, P. L. Pauson and C. F. Farnocchi, J. Organomet. Chem., 1988, 356, 213–219 CrossRef CAS; (b) J. K. Whitesell, M. A. Minton and S. W. Felman, J. Org. Chem., 1983, 48, 2193–2195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR and HPLC data. See DOI: https://doi.org/10.1039/d2cc03648g |
| This journal is © The Royal Society of Chemistry 2022 |