
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHuman endonuclease III/NTH1: focusing on the [4Fe–4S] cluster and the N-terminal domain†
Elin
Moe
 a,
Célia M.
Silveira
a,
Lidia
Zuccarello
a,
Filipe
Rollo
a,
Meike
Stelter
b,
Salvatore
De Bonis
b,
Catharina
Kulka-Peschke
c,
Sagie
Katz
c,
Peter
Hildebrandt
c,
Ingo
Zebger
c,
Joanna
Timmins
*b and
Smilja
Todorovic
*a
a,
Célia M.
Silveira
a,
Lidia
Zuccarello
a,
Filipe
Rollo
a,
Meike
Stelter
b,
Salvatore
De Bonis
b,
Catharina
Kulka-Peschke
c,
Sagie
Katz
c,
Peter
Hildebrandt
c,
Ingo
Zebger
c,
Joanna
Timmins
*b and
Smilja
Todorovic
*a
aInstituto de Tecnologia Química e Biológica António Xavier, Universidade NOVA de Lisboa, Av. da República, 2780-157, Oeiras, Portugal. E-mail: smilja@itqb.unl.pt
bUniv. Grenoble Alpes, CEA, CNRS, IBS, F-38000, Grenoble, France. E-mail: joanna.timmins@ibs.fr
cInstitut für Chemie, Sekr. PC14, Technische Universität Berlin, Straße des 17. Juni 135, D-10623, Berlin, Germany
First published on 24th October 2022
Abstract
Human Endonuclease III (EndoIII), hNTH1, is an FeS containing enzyme which repairs oxidation damaged bases in DNA. We report here the first comparative biophysical study of full-length and an N-terminally truncated hNTH1, with a domain architecture homologous to bacterial EndoIII. Vibrational spectroscopy, spectroelectrochemistry and SAXS experiments reveal distinct properties of the two enzyme forms, and indicate that the N-terminal domain is important for DNA binding at the onset of damage recognition.
hNTH1 is crucial for genome maintenance in humans, as the mutations in the nth1 gene are associated with development of adenomatous polyposis and colorectal cancer.1 Primary sequence comparison of human and bacterial EndoIIIs reveals that the enzymes share similar catalytic domains, but that hNTH1 possesses an additional N-terminal domain (NTD) consisting of approximately 90 amino acids (Fig. S1, ESI†). The role of the NTD is not fully understood but has been linked to nuclear and mitochondrial targeting and/or interactions with other base excision repair (BER) and non-BER proteins.2–5 Recently the three-dimensional structure of hNTH1 lacking the first 63 amino acids (hNTH1Δ63) was determined.6 In this structure the first twenty amino acids are not visible in the electron density map, indicating a distinct flexibility of the NTD. Overall, the domain composition and organization of hNTH1Δ63 are similar to that of bacterial EndoIIIs,7–9 however, domains A and B adopt an ‘open’ configuration in hNTH1Δ63, which is distinct from the ‘closed’ bacterial EndoIIIs (Fig. S2, ESI†). In the ‘open’ conformation, the distance between the two catalytic residues (Lys220 and Asp239) is more than 23 Å, which are thus not positioned to process DNA (Fig. S2, ESI†). The unique conformation of hNTH1Δ63 was attributed to a more flexible linker connecting domains A and B, which when substituted by the shorter linker found in E. coli EndoIII (EcEndoIII) resulted in a ‘closed’ conformation.6 The transition from an ‘open’ to a ‘closed’ conformation was proposed to be induced upon DNA binding (between domain A and B), as observed in a DNA-bound bacterial EndoIII.8 The role of the [4Fe–4S] cluster in EndoIII enzymes is still under debate. In early studies of bacterial EndoIIIs, it was proposed to play a structural role, due to lack of redox activity in solution.10,11 More recently, it has been suggested to be involved in a redox transition related DNA damage search and signalling processes, in either a DNA-dependent12 or DNA-independent way.13 Here, we report the structural and redox properties of the cluster found in hNTH1 and an N-terminally truncated hNTH1 that lacks the first 89 amino acids (hNTH1Δ89). The interaction of the two enzymes with DNA substrates was probed by vibrational spectroscopy, while the conformational flexibility was evaluated by small-angle X-ray scattering (SAXS) and AlphaFold models.14 The results are discussed in light of reports on bacterial EndoIII enzymes and the recent crystal structure of hNTH1Δ63.
Resonance Raman (RR) spectra, which are sensitive to the cluster type, structure and symmetry, indicate a fingerprint of a typical fully cysteinyl coordinated [4Fe–4S]2+ cluster in hNTH1.15 The most prominent RR bands at 335, 358, 365 and 388 cm−1 are attributed to bridging, (Fe–S)b, (335 and 388 cm−1) and terminal, (Fe–S)t, (358 and 365 cm−1) modes of the cubane structure, involving inorganic S atoms and Cys S atoms, respectively (Fig. S3, ESI†). The spectra of hNTH1 are very similar to those reported for EcEndoIII11 and Deinococcus radiodurans EndoIII variant 2 (DrEndoIII2)13 and are almost indistinguishable from RR spectra of DrEndoIII variant 3 (DrEndoIII3) (Fig. S3, ESI†). No changes could be observed in UV-vis spectra of hNTH1 in solution upon addition of oxidizing or reducing agents, as previously observed for bacterial EndoIIIs (Fig. S4, ESI†).10,13,16 Next, to investigate the redox properties of their [4Fe–4S] clusters, hNTH1 and hNTH1Δ89 were immobilized, as reported previously for bacterial EndoIIIs,12,13 on biocompatible Au electrodes coated with either carboxyl-, undamaged DNA- or damaged DNA-terminated self-assembled monolayers (SAMs). The latter carries the thymine glycol (Tg) oxidized base, a well-established substrate of hNTH1 and EndoIII enzymes. The adsorption of the proteins onto the biocompatible electrodes was monitored by Surface Enhanced Infrared Absorption (SEIRA) spectroscopy, which enhances the IR absorption intensity of molecules close to the surface by up to two orders of magnitude.13 Amide I (centred at 1660 cm−1) and amide II (centred at 1550 cm−1) bands, originating from the protein backbone, were detected for both enzymes on mercaptoundecanoic acid (MUA) and damaged DNA SAMs (Fig. 1A and B). Importantly, unlike hNTH1Δ89 and bacterial EndoIIIs, hNTH1 does not bind to electrodes coated with undamaged DNA (Fig. 1A). Also, the amide I band normalized SEIRA spectra of hNTH1Δ89, attached to different SAMs do not display any significant differences (Fig. S5A, ESI†). Interestingly, the ratios of the amide I and amide II band intensities in SEIRA spectra of hNTH1 and hNTH1Δ89, which are indicative of the orientation of the enzyme helices with respect to the electrode surface,13 are comparable regardless of the SAM (i.e. MUA and damaged DNA) (Fig. S5B, ESI†). Note that SEIRA band intensity is higher for MUA than for DNA SAMs, indicating a lower number of attached enzyme molecules, which could be due to less compact packing (ESI†) and/or a lower affinity of enzyme molecules in the case of the latter.
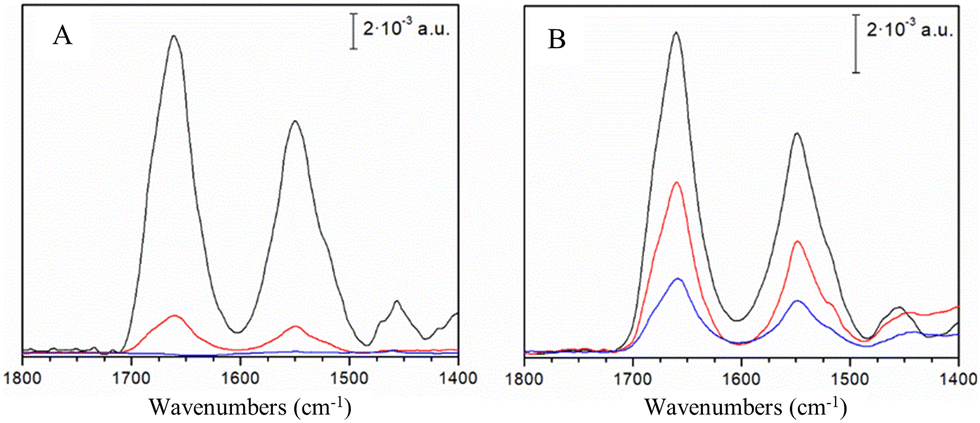 | ||
| Fig. 1 SEIRA spectra of hNTH1 and hNTH1Δ89. (A) hNTH1 and (B) hNTH1Δ89 adsorbed on a gold electrode surface coated with MUA (black), damaged DNA (red) and undamaged DNA (blue) SAMs. | ||
Among the tested SAM/enzyme electrode-constructs, only hNTH1Δ89 immobilized on MUA exhibits faradaic CV peak currents (Fig. S6A, ESI†), with a midpoint potential (Em) of 38 ± 5 mV vs. NHE. By analogy with DrEndoIII2, we attribute it to the +2/+1 transition of the [4Fe–4S]2+cluster.13 The obtained value is in the same range as those reported for EcEndoIII (58 mV)12 and DrEndoIII2 (20 mV),13 and lower than the values reported for A. fulgidus uracil DNA glycosylase (95 mV) and E. coli MutY (90 mV).12 The linear peak current dependence of the scan rate (Fig. S6B, ESI†) confirms a surface confined electron transfer (ET) process, with rate constant (kET) of 8.7 ± 0.1 s−1, which is comparable to those reported for bacterial EndoIIIs.12,13 These findings support the growing evidence that redox activation of EndoIIIs is not necessarily triggered by the presence of DNA, but instead may be facilitated by strong electrostatic interactions, possibly with partner proteins as suggested previously.13 Further investigations of the in vivo molecular interactions involving hNTH1 will be needed to corroborate this hypothesis. The scenario is probably even more complex, since, unlike its bacterial counterparts, the cluster in hNTH1 does not undergo redox transition upon attachment of the enzyme to electrodes modified with MUA. Furthermore hNTH1, unlike its truncated form and bacterial homologues, does not interact at all with undamaged DNA SAMs (Fig. 1A).
SAXS experiments performed on hNTH1 and hNTH1Δ89 in either absence or presence of damaged DNA (carrying tetrahydrofuran, THF, moiety, ESI†) reveal that only hNTH1 undergoes a remarkable conformational change upon DNA binding (Table 1 and Fig. S7B, Table S1, ESI†). This can be seen as a marked reduction in the radius of gyration (Rg) and the maximal particle dimension (Dmax) of the complex compared to the unbound enzyme (Table 1 and Fig. S7C, ESI†). In contrast, and as expected for a complex versus the enzyme alone, the I(0) and Porod volumes that reflect the molecular weight of the particles in solution, are higher for the complex compared to hNTH1 alone (Table 1). The shape of the hNTH1 SAXS curve in the presence of DNA is also significantly modified (Fig. S7A, ESI†). In the case of hNTH1Δ89, DNA binding also leads to a change in the shape of the SAXS curve, but the reduction in the Rg and Dmax is less pronounced (Table 1). The larger changes in Rg and Dmax observed in hNTH1 upon DNA binding are thus likely related, in part at least, to the flexible NTD.
| Sample | Mean Rg (nm) | I(0) | Porod vol. (Å3) | D max |
|---|---|---|---|---|
| hNTH1 | 2.93 ± 0.02 | 11.67 ± 0.030 | 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 241 241 |
7.93 |
| hNTH1-dsDNA-THF | 2.18 ± 0.01 | 21.46 ± 0.016 | 48677 |
7.12 |
| hNTHΔ89 | 2.23 ± 0.01 | 5.39 ± 0.016 | 33208 |
5.55 |
| hNTHΔ89-dsDNA-THF | 2.03 ± 0.01 | 13.03 ± 0.006 | 42648 |
5.17 |
AlphaFold14 models of hNTH1 are in good agreement with the SAXS data, and strongly indicate that the NTD is highly disordered and mostly extended, adopting a wide range of conformations (Fig. 2A and B). Both the Rg and Dmax values of hNTH1 are indeed much higher than those of hNTH1Δ89 (Table 1). In contrast, upon DNA binding, it appears that the NTD wraps tightly around the catalytic domain and the DNA, resulting in a much more compact structure, similar to the ‘closed’ hNTH1Δ89 conformation bound to DNA (Fig. 2C).
 | ||
| Fig. 2 Conformational flexibility of hNTH1 and hNTH1Δ89 in solution. Crysol derived fits of the experimental SAXS scattering curves (blue) to the theoretical scattering curves (red). (A–C) hNTH1 vs. AlphaFold derived model of (A) ‘closed’ and (B) ‘open’ state hNTH1, (C) hNTH1 bound to DNA vs. ‘closed’ model of hNTH1Δ63 (PDB, 7RDS) bound to dsDNA (extracted from PDB 1P59). (D–F) hNTH1Δ89 vs. (D) ‘open’ and (E) ‘closed’ state of hNTH1Δ63; and (F) hNTH1Δ89 bound to DNA vs. ‘closed’ state of hNTH1Δ63 bound to dsDNA. Chi2 values (goodness of fit) are indicated. The respective atomic models used for the fits are illustrated in rainbow from N- to C-terminus for the AlphaFold models and in blue (protein) and beige (DNA) models of hNTH1Δ63. Positively charged residues (Arg and Lys) located in the flexible NTD are shown as sticks in (A) and (B). | ||
To determine whether hNTH1 adopts an ‘open’ or a ‘closed’ conformation in solution, we generated seven high-confidence AlphaFold models of hNTH1 in either ‘closed’ (five models) or ‘open’ (two models) states (ESI†) and fitted the theoretical curves of these models to the experimental hNTH1 SAXS data (Fig. 2A and B). All models presented extended and unfolded NTDs, in agreement with the large Rg and Dmax values of DNA-free hNTH1. Using Crysol, we fitted the theoretical curves of each of these models to the experimental SAXS data (Fig. 2A and B). These fits clearly indicate that hNTH1 adopts preferentially a ‘closed’ conformation in solution (Fig. 2A). Two of the ‘closed’ conformation models produced fits with Chi2 values under 1 (the other three models gave Chi2 values between 2.5 and 10), whereas the quality of the fits to the two ‘open’ conformation models were poorer with Chi2 values of 10.8 and 17.9 (Fig. 2B). In contrast, the fitting of the hNTH1Δ89 SAXS data to theoretical curves of the crystal structures of hNTH1Δ63 in either ‘open’ or ‘closed’ conformation (Fig. 2D and E) confirms that hNTH1Δ89 indeed adopts an ‘open’ conformation in which domain A is rotated away from domain B (Fig. 3A) as reported recently in the crystal structure of hNTH1Δ63.6 Carroll et al. proposed that this alternative conformation may relate to the longer linker region connecting domains A and B in hNTH1 compared to its bacterial homologues.6 However, this model is not supported by our finding that the full-length enzyme, hNTH1, which also bears a longer linker, adopts a ‘closed’ conformation (Fig. 2A). Instead, we propose that the ‘open’ form adopted by hNTH1Δ63 (and hNTH1Δ89) may not be physiologically relevant and may result from the absence of the NTD and the presence of the flexible linker. On the contrary, the ‘closed’ conformation of hNTH1 may be stabilized by interactions between the end of the NTD and the catalytic domain, in particular domain A (Fig. 3A). Interestingly, upon DNA binding, this conformational flexibility is lost, and both enzyme forms adopt similar, ‘closed’ conformations (Fig. 2C, F and 3B, C). Rotation of domain A relative to domain B needed for DNA binding thus only occurs in hNTH1Δ89, while the full-length enzyme appears to be in a ‘closed’ state in absence/presence of DNA.
 | ||
| Fig. 3 Schematic representation of orientations and conformations of hNTH1 (left) and hNTH1Δ89 (right). (A) Models of hNTH1 and hNTH1Δ89 in DNA free states, based on the fitting of SAXS data to experimentally derived and predicted structures. (B) Damaged-DNA terminated SAMs: both hNTH1 and hNTH1Δ89 adopt a ‘closed’ conformation and the NTD of hNTH1 wraps tightly around the protein/DNA. (C) Undamaged-DNA terminated SAMs: only hNTH1Δ89 binds to the SAM, most likely in a ‘closed’ conformation. (D) MUA-terminated SAMs: hNTH1 and hNTH1Δ89 bind to the SAM in different orientations, suggesting that the NTD bearing positively charged residues plays a role in binding to negatively charged MUA. | ||
We provide here evidence that hNTH1 is distinct from NTD-truncated constructs and bacterial EndoIIIs in many regards. This is particularly relevant since the current mechanistic models are largely derived from the studies of bacterial homologues. Firstly, the full-length enzyme does not interact with undamaged DNA, which represents one of the major cornerstones of the currently proposed mechanistic model of EndoIIIs.12 This finding may denote that the NTD is involved in discrimination between undamaged and damaged DNA. Further investigation will be needed to verify this hypothesis. hNTH1 has in fact previously been reported to exhibit low affinity (μM range) binding to undamaged DNA and high affinity (nM range) binding to its damaged DNA substrate in biochemical assays.17 In the present study, the SAXS data also suggest that the positively charged NTD may be directly involved in DNA binding by wrapping tightly around the catalytic domain and DNA substrate. Secondly, SEIRA spectra of hNTH1 and hNTH1Δ89 indicate comparable orientations of the proteins relative to the damaged DNA-terminated SAM. This is also supported by the SAXS data that suggests that both hNTH1 and hNTH1Δ89 adopt a ‘closed’, compact conformation with the DNA duplex bound in the cleft between domains A and B. The observed lack of redox activity of hNTH1 upon attachment to MUA coated electrodes could be the consequence of either an unfavourable orientation of the immobilized protein with respect to the electrode or a large distance between the [4Fe–4S] cluster and the electrode surface preventing efficient heterogeneous ET. Due to comparable amide I and amide II band ratios measured for hNTH1 and hNTH1Δ89 attached to MUA, we hypothesize that the negatively charged MUA may potentially interact with the positively charged NTD in hNTH1, thereby increasing the distance between the electrode surface and the cluster (Fig. 3D). The redox transition of hNTH1Δ89 observed upon attachment to MUA may in contrast result from the interaction between MUA and a positively charged patch close to the [4Fe–4S] cluster that is more accessible in the truncated form (Fig. S2, ESI†).
In conclusion, we provide direct spectroscopic evidence that the previously suggested DNA mediated ET role of the FeS clusters of EndoIII enzymes, may not be valid for hNTH1, as it does not bind to undamaged DNA. Instead, we propose that the FeS cluster plays a role as a structural scaffold which is critical for stabilizing the interaction with DNA upon damage recognition, as previously suggested for MutY.18 Further, we demonstrate that hNTH1 and hNTH1Δ89 possess distinct DNA binding properties, with hNTH1 undergoing a major conformational change upon binding to damaged DNA that notably involves its flexible NTD. Altogether, our spectroscopic and SAXS data highlight important biophysical differences between full-length and truncated hNTH1, which implies that mechanistic insights provided by studies of bacterial homologues or truncated forms of hNTH1 may not be applicable to the physiologically relevant human protein.
E. M.: writing – original draft, C. M. S.: writing – review & editing, L. Z., F. R., M. S., S. B. and C. K.-P.: investigation, S. K., P. H. and I. Z.: writing – review & editing, funding acquisition, J. T. and S. T.: conceptualization, writing – original draft, funding acquisition.
We thank M. Brennich for assistance on the ESRF SAXS beamline and F. Dehez for advice with AlphaFold predictions. Funding is acknowledged from FCT – through MOSTMICRO-ITQB R&D Unit (UIDB/04612/2020, UIDP/04612/2020) and LS4FUTURE Associated Laboratory (LA/P/0087/2020); SFRH/BD/132966/2017; Comité de l’Isère de La Ligue Contre le Cancer (Project R21Timmins); ARC (PJA 20151203255); la Fondation de France (MS: 2012-00034094); DFG-EXC 2008-390540038-UniSysCat; SPP 1927 “Iron–sulfur for Life” 10043725 (ZE 510/2-1), CEA Radiobiology Program and EU's Horizon 2020 research and innovation programme (grant agreement No 810856).
Conflicts of interest
There are no conflicts to declare.References
- R. D. A. Weren, M. J. L. Ligtenberg, A. Geurts van Kessel, R. M. De Voer, N. Hoogerbrugge and R. P. Kuiper, J. Pathol., 2018, 244, 135–142 CrossRef PubMed.
- S. Ikeda, T. Biswas, R. Roy, T. Izumi, I. Boldogh, A. Kurosky, A. H. Sarker, S. Seki and S. Mitra, J. Biol. Chem., 1998, 273, 21585–21593 CrossRef CAS PubMed.
- M. L. Hegde, T. K. Hazra and S. Mitra, Cell. Mol. Life Sci., 2010, 67, 3573–3587 CrossRef CAS PubMed.
- M. Fuxreiter, P. Tompa, I. Simon, V. N. Uversky, J. C. Hansen and F. J. Asturias, Nat. Chem. Biol., 2008, 4, 728–737 CrossRef CAS PubMed.
- P. Tompa and M. Fuxreiter, Trends Biochem. Sci., 2008, 33, 2–8 CrossRef CAS PubMed.
- B. L. Carroll, K. E. Zahn, J. P. Hanley, S. S. Wallace, J. A. Dragon and S. Doublié, Nucleic Acids Res., 2021, 49, 13165–13178 CrossRef CAS PubMed.
- M. M. Thayer, H. Ahern, D. Xing, R. P. Cunningham and J. A. Tainer, EMBO J., 1995, 14, 4108–4120 CrossRef CAS PubMed.
- J. C. Fromme and G. L. Verdine, EMBO J., 2003, 22, 3461–3471 CrossRef CAS PubMed.
- A. Sarre, M. Ökvist, T. Klar, D. Hall, A. Smalas, S. McSweeney, J. Timmins and E. Moe, J. Struct. Biol., 2015, 191, 87–99 CrossRef CAS PubMed.
- R. P. Cunningham, H. Asahara, J. F. Bank, C. P. Scholes, J. C. Salerno, K. Surerus, E. Munck, J. McCracken, J. Peisach and M. H. Emptage, Biochemistry, 1989, 28, 4450–4455 CrossRef CAS PubMed.
- W. Fu, S. O’Handley, R. P. Cunningham and M. K. Johnson, J. Biol. Chem., 1992, 267, 16135–16137 CrossRef CAS PubMed.
- A. K. Boal, E. Yavin, O. A. Lukianova, V. L. O’Shea, S. S. David and J. K. Barton, Biochemistry, 2005, 44, 8397–8407 CrossRef CAS PubMed.
- E. Moe, M. Sezer, P. Hildebrandt and S. Todorovic, Chem. Commun., 2015, 51, 3255–3257 RSC.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- S. Todorovic and M. Teixeira, J. Biol. Inorg. Chem., 2018, 23, 647–661 CrossRef CAS PubMed.
- G. Caserta, L. Zuccarello, C. Barbosa, C. M. Silveira, E. Moe, S. Katz, P. Hildebrandt, I. Zebger and S. Todorovic, Coord. Chem. Rev., 2022, 452, 214287 CrossRef CAS.
- A. Sarre, M. Stelter, F. Rollo, S. De Bonis, A. Seck, C. Hognon, J. L. Ravanat, A. Monari, F. Dehez, E. Moe and J. Timmins, DNA Repair, 2019, 78, 45–59 CrossRef CAS PubMed.
- M. P. Golinelli, N. H. Chmiel and S. S. David, Biochemistry, 1999, 38, 6997–7007 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, Fig. S1–S9 and Table S1. See DOI: https://doi.org/10.1039/d2cc03643f |
| This journal is © The Royal Society of Chemistry 2022 |