
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deracemisation and stereoinversion by a nanoconfined bidirectional enzyme cascade: dual control by electrochemistry and selective metal ion activation†
Beichen
Cheng
 a,
Rachel S.
Heath
b,
Nicholas J.
Turner
b,
Fraser A.
Armstrong
a and
Clare F.
Megarity‡
*a
a,
Rachel S.
Heath
b,
Nicholas J.
Turner
b,
Fraser A.
Armstrong
a and
Clare F.
Megarity‡
*a
aDepartment of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: clare.megarity@manchester.ac.uk
bSchool of Chemistry, University of Manchester, Manchester Institute of Biotechnology, 131 Princess Street, Manchester, M1 7DN, UK
First published on 30th September 2022
Abstract
The unique ability of the ‘electrochemical leaf’ (e-Leaf) to drive and control nanoconfined enzyme cascades bidirectionally, while directly monitoring their rate in real-time as electrical current, is exploited to achieve deracemisation and stereoinversion of secondary alcohols using a single electrode in one pot. Two alcohol dehydrogenase enzymes with opposing enantioselectivities, from Thermoanaerobacter ethanolicus (selective for S) and Lactobacillus kefir (selective for R) are driven bidirectionally via coupling to the fast and quasi-reversible interconversion of NADP+/NADPH catalysed by ferredoxin NADP+ reductase – all enzymes being co-entrapped in a nanoporous indium tin oxide electrode. Activity of the Lactobacillus kefir enzyme depends on the binding of a non-catalytic Mg2+, allowing it to be switched off after an oxidative half-cycle, by adding EDTA – the S-selective enzyme, with a tightly-bound Zn2+, remaining fully active. Racemate → S or R → S conversions are thus achieved in high yield with unprecedented ease.
The ‘electrochemical leaf’ (e-Leaf) greatly extends protein film electrochemistry to include the ability to drive and control enzymes lacking an electron-transfer role. In the e-Leaf, ferredoxin NADP+ reductase (FNR) is bound tightly at high local concentration in the nanocavities of a highly porous indium tin oxide (ITO) layer formed on a conducting support. Under potential control, fast electron exchange between its active site flavin and the electrode results in the rapid bidirectional interconversion of NADP+/NADPH which is immediately coupled to a co-entrapped NADP(H)-dependent dehydrogenase.1–3 Extended enzyme cascades can also be loaded into the electrode pores where they are driven via this coupling which requires only one of the enzymes to be NADP(H)-dependent.4 The crowded environment mimics the nanoconfinement of metabolic enzyme cascades in biology. The unique combination – retention of intermediates and cofactors, the ability to energise and to control the direction and rate of the reaction, combined with real-time monitoring (which allows immediate observation of the response to interventions, such as addition5 or removal by, for example, replacement of the bulk solution2 or simply by chelation,6 of an effector) has already been exploited in several ways.5,7,8 Examples include the energisation and control of an extended five-enzyme cascade,4 monitoring of slow binding drugs to isocitrate dehydrogenase,6 and a two-step deracemiser.9 Most recently, a four-enzyme cascade, simultaneously driven by electrical and chemical energy, incorporated in situ ATP-recycling.10 The e-Leaf electrode is represented by the notation (E1 + E2…)@ITO/support, where E1 = FNR, E2 is a dehydrogenase and the support is graphite or titanium foil (the latter allowing double-sided electrophoretic deposition of ITO).
Most molecules in living organisms are chiral and therefore the binding of a drug is highly enantiomer-specific, exemplified by Thalidomide.11–13 For the pharmaceutical industry, effective deracemisation is therefore paramount. Our previous use of the e-Leaf for deracemisation9 sought to overcome compatibility problems encountered in common approaches.14–22 An alcohol dehydrogenase (ADH) pair with opposing enantioselectivities was used to resolve a racemic mixture in two steps.9 This work revealed an important consequence of rapid and reversible enzyme reactions occurring in the nanopore environment: local equilibrium is quickly attained, thereby favouring racemisation rather than deracemisation. A two-electrode system was thus needed to separate the oxidation and reduction stages.9
Here, we present a new strategy for deracemisation and stereoinversion, which exploits the different metal ion selectivities of two alcohol dehydrogenases loaded into the ITO electrode. Activity of the R-specific enzyme conveniently depends on the binding of Mg2+ ions, which allows it to be switched off at the half-way stage of an oxidation/reduction cycle, simply by adding EDTA; the other ADH in the pair (the active site of which contains a tightly-bound Zn2+) remaining unaffected (ESI,† Section S5). With this dual control, deracemisation or stereoinversion at high yield are thus achievable in the simplest way, a one-pot, one-electrode electrolyser.
Scheme 1 outlines the concept: FNR and an S-selective and R-selective ADH are nanoconfined in the porous electrode. When starting with a racemic mixture of alcohol, an oxidising potential is applied to drive conversion to the ketone, each enantiomer being processed by its respective ADH. The progress of the reaction is monitored until the current approaches zero and the charge passed equates to the amount of ketone produced. At this point, the R-selective ADH is de-activated by adding EDTA to chelate Mg2+, the electrode thus becoming strongly S-selective. The potential is then reversed to drive the reduction of the ketone back to S-alcohol, by the S-selective ADH, thus resolving the racemic mixture. An analogous approach is taken to undertake the stereoinversion, R → S.
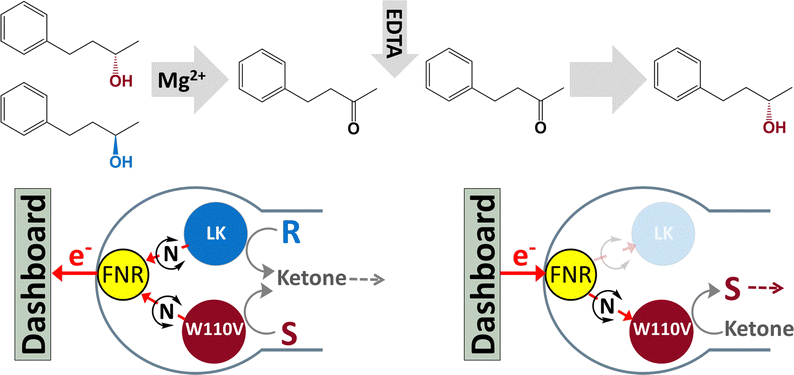 | ||
| Scheme 1 Deracemisation by dual control of ADH enzymes with opposing enantioselectivities, nanoconfined and driven in the porous electrode of the e-Leaf. The dashboard represents the potentiostat and computer for potential control and real-time monitoring. (For stereoinversion, the starting racemic mixture is replaced with R-alcohol.) | ||
The important differences between the two ADHs are highlighted in Fig. 1. The S-selective variants which derive from Thermoanaerobacter ethanolicus, are homo-tetramers, each monomer containing a tightly bound catalytic Zn2+. The tryptophan at position 110 is exchanged for either alanine or valine, the latter having superior S-selectivity (they are referred to as W110A and W110V throughout).23,24 The R-selective enzyme (referred to as LK) which is also a homo-tetramer, derives from Lactobacillus kefir. This enzyme uses a catalytic triad instead of Zn2+ at the active site, but exhibits a strong dependency on non-catalytic Mg2+ for activity25,26 Hypotheses on the role of Mg2+ are outlined in Section S2 ESI,†.
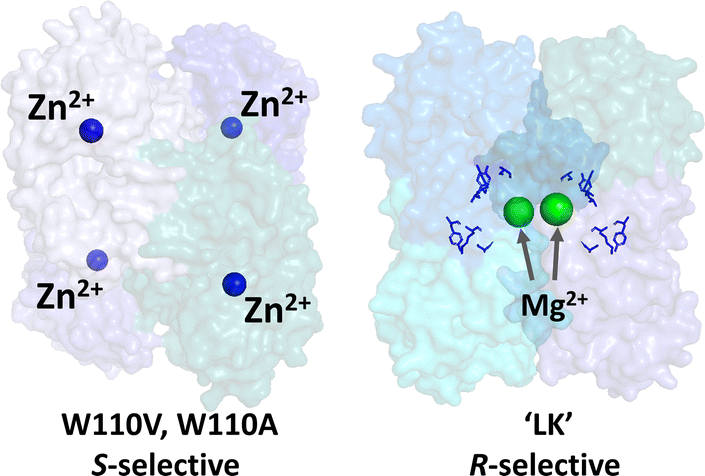 | ||
| Fig. 1 Alcohol dehydrogenases with opposing enantioselectivities. Left: S-selective ADH from Thermoanaerobacter ethanolicus, catalytic Zn2+ shown as blue spheres (PDB 7JNS). Right: R-selective ADH from Lactobacillus brevis, structural Mg2+ ions shown as green spheres and the catalytic triad residues, Ser-Tyr-Lys, shown in blue; this enzyme lacks Zn2+ (PDB 6H07). | ||
Table 1 lists the kinetic data and enantiomeric preferences for each enzyme used in this study. The LK enzyme has no measurable activity with S-alcohols. Its high R-selectivity arises because during a catalytic cycle, the pro-R hydride from the nicotinamide is transferred to the si face of the carbonyl to give R-alcohols. Rotation of a bound ketone around its carbonyl bond such that its re face would be positioned to accept the hydride, would result in extreme steric hinderance.25,27 The relatively high S-selectivity of W110A is increased further by incorporating the bulkier residue, valine. This mutation renders the enzyme inactive for R- but also less active for S-alcohols.
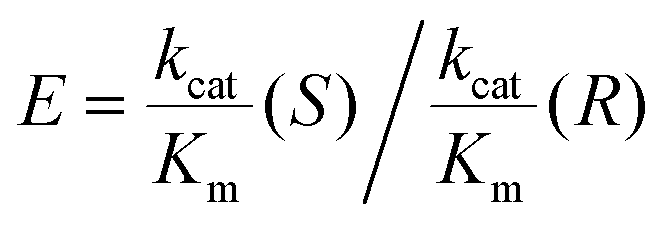
Fig. 2 shows the stereoinversion of (R)-4-phenyl-2-butanol contained initially at a concentration of 4 mM in 5 mL of aqueous solution (20 μmoles). The (FNR + LK + W110V)@ITO/Ti foil electrode (7 cm2) was prepared by preloading with FNR (drop-casting a concentrated solution) after which LK and W110V were loaded from a dilute solution containing 1.28 nmoles of LK and 0.66 nmoles of W110V. The electrode was rinsed thoroughly before use to remove unbound enzymes. The starting solution (pH 9.0, chosen to favour oxidation during the first stage9) also contained 5 mM Mg2+. Holding at a potential of +0.21 V vs. SHE, the oxidation half-cycle was initiated by introducing NADP+ to a final concentration of 20 μM, which resulted in a rapid increase in oxidation current attributable to the conversion of (R)-4-phenyl-2-butanol to 4-phenyl-2-butanone by LK. After approximately 24 h, when the current had decreased to a low level, the potential was set to open circuit and a sample was analysed by chiral GC. The result showed (Table S3 ESI,†) that 85.7% of the alcohol had been converted to ketone, the remainder being unreacted R- and a small amount of S-alcohol. The charge passed during the oxidation phase equated to the conversion of 17.0 μmoles (equivalent to 85% of the starting alcohol, in good agreement with the GC analysis which showed a conversion of 17.14 μmoles). The total number of turnovers undergone by each (averaged) NADP+ at this stage was 170.
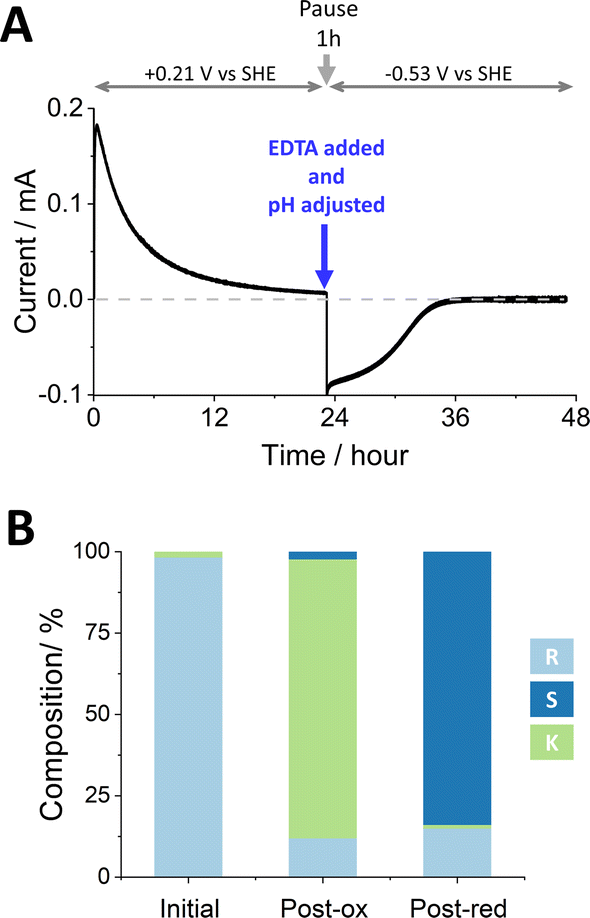 | ||
| Fig. 2 The stereoinversion of (R)-4-phenyl-2-butanol by W110V and LK nanoconfined in the e-Leaf. (A) As monitored by chronoamperometry following the addition of NADP+ at t = 0. At the end of the oxidation phase, a 1 mL sample was removed for chiral GC analysis and by a single addition, the pH was adjusted to 8 and EDTA was introduced to chelate Mg2+ thus inactivating LK (the reaction was paused for 1 h to ensure complete chelation). The potential was then switched to drive reduction, and measurement resumed. (B) Bulk solution compositions analysed by chiral GC. Conditions: initial cell volume 5 mL; solution stirred by magnetic flea; substrate [(R)-4-phenyl-2-butanol] = 4 mM, [NADP+] = 20 μM, [Mg2+] = 5 mM; buffer: 50 mM TAPS pH 9.0 at the start, adjusted to 8 after phase one; temperature: 25 °C; potential (vs. SHE) held at +0.21 V for oxidation and −0.53 V for reduction. An anaerobic glovebox was used to avoid contribution to the current from reduction of O2. Enzyme loading: FNR loaded first by drop-casting, LK and W110V loaded after FNR, by stirring in a 2.5 mL solution containing 1.28 nmols of LK and 0.66 nmols of W110V in 50 mM TAPS buffer pH 9; electrode (ITO@Ti): 7 cm2. | ||
To prepare for the reduction phase, EDTA was added (to give a final concentration of 25 mM) and the pH was adjusted to 8.0 (to favour reduction9). After a pause lasting 1 h to ensure complete chelation of Mg2+ and inactivation of LK (the EDTA causes release of Mg2+ from LK but not release of the Zn2+ from W110V), an electrode potential of −0.53 V vs. SHE was applied, resulting in a rapid increase in reduction current which gradually decreased to zero after approximately 12 h. The total charge passed during the reduction phase equated to the conversion of 11.4 μmoles of ketone. Analysis of the main cell solution by chiral GC showed 2.78 mM (S)-4-phenyl-2-butanol, and 0.50 mM (R)-4-phenyl-2-butanol, corresponding to 11.1 and 2.0 μmoles, respectively. The solution in the side arm was also analysed by chiral GC to quantify material that had ‘crossed over’ through the porous frit. The combined data are shown in Table S3 (ESI†), which includes % conversion at each step along with ee% values. The total number of turnovers undergone by each (averaged) NADP+ throughout the two stages was 313.
Fig. 3 shows an experiment to undertake the deracemisation of an 8 mM solution of (±)-4-phenyl-2-butanol. The (FNR + LK + W110V)@ITO/Ti foil electrode (14 cm2 total area) was prepared as for the stereoinversion experiment shown in Fig. 2, and the solution, at pH 9.0, contained 5 mM Mg2+. Upon initiation of the oxidation phase by addition of NADP+, there was a rapid increase in current which gradually approached zero after approximately 40 h. The longer time taken compared to stereoinversion is explained by the inherently slow rate of W110V, required for deracemisation but inconsequential to the stereoinversion experiment where only LK is required for oxidation of the starting R-alcohol substrate.
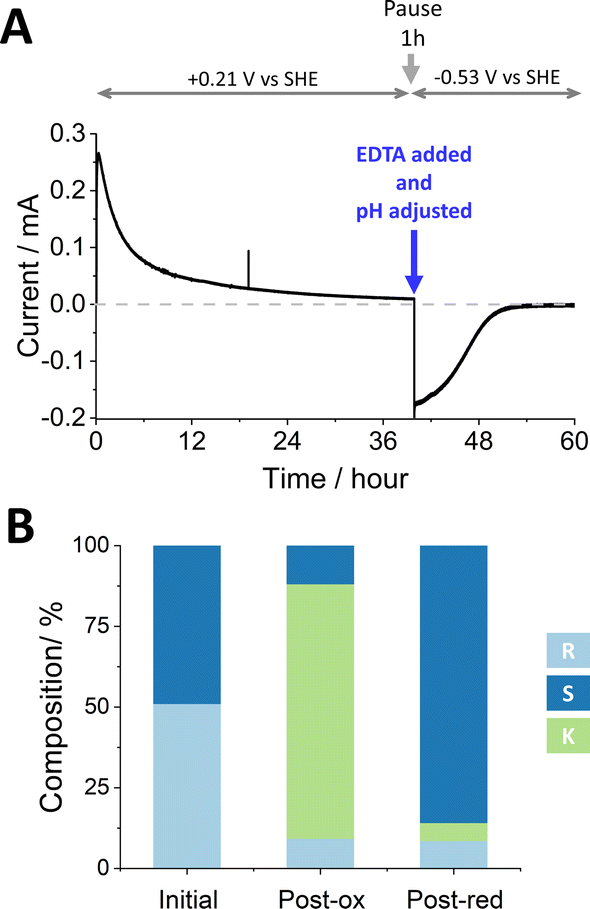 | ||
| Fig. 3 Deracemisation of a racemic mixture of (±)-4-phenyl-2-butanol by nanoconfined W110V and LK. (A) The reaction course monitored by chronoamperometry. Experimental steps carried out as for Fig. 1. (B) Bulk solution composition analysed by chiral GC. Conditions: as for Fig. 2 apart from starting substrate: [(±)-4-phenyl-2-butanol] = 8 mM and amount of ADH enzymes: 2.58 nmols of LK and 1.31 nmols of W110V; electrode (ITO@Ti): 14 cm2. | ||
The charge passed during the oxidation phase equated to the conversion of 33.4 μmoles of reactant (83.5% of the quantity of (±)-4-phenyl-2-butanol). For comparison, GC analysis showed 4-phenyl-2-butanone at a concentration of 6.31 mM (31.6 μmoles; 78.9% conversion). The 5% discrepancy is explained in part by the crossover of ketone into the side arm which was not analysed by chiral GC at the midway point. Despite the current approaching zero, oxidation was incomplete since both R- and S-alcohols were still present at 0.73 mM and 0.95 mM respectively (Fig. 3B). We return to this result later.
The reduction phase was carried out as for Fig. 2, after injecting EDTA to give a final concentration of 25 mM, adjusting the pH to 8.0, and applying a potential of −0.53 V vs. SHE after a 1 h pause. The current dropped to zero after approximately 10 h. The total charge passed during this phase equated to a conversion of 21.1 μmoles. Analysis of the main cell solution by chiral GC showed 5.06 mM (S)-4-phenyl-2-butanol, and 0.50 mM (R)-4-phenyl-2-butanol, corresponding to 20.2 and 2.0 μmoles, respectively. The side-arm solution was also analysed. The combined data for this experiment are shown in Table S4 (ESI†), which includes conversions to % conversion at each step along with ee% values. The total number of turnovers undergone by each (averaged) NADP+ throughout the two stages was 598.
Equivalent experiments using W110A are shown in Section S3, ESI.† The stereoinversion experiment using this variant gave an inferior outcome overall, with more ketone remaining (11.3% vs. 1.1%) and less S-alcohol (75.5% vs. 83.9%). As shown in Fig. 2, use of the W110V variant, optimising for the reduction by pH adjustment midway, and ensuring complete inactivation of LK by prolonged chelation time, resulted in a better outcome. For deracemisation, any distinction between W110A and W110V was less clear. Despite having slightly more S-alcohol (86% vs. 84%) and less ketone (5.6% vs. 9.5%) in the final solution, use of W110V resulted in a similar enantiomeric excess to that achieved with W110A (ee%: 82 vs. 85.7). In all experiments, the R-enantiomer persisted to some extent. As proposed previously, an important factor may be the crowded nanoporous environment that traps reactants and products for sufficient time to accelerate the approach to final equilibrium, which always favours a racemic mixture.9
In this proof-of-concept, we have exploited, (i) the ability to drive ADH enzymes with opposing enantioselectivities, bidirectionally, (ii) the ability to inactivate, selectively, the R-specific enzyme, (iii) the ability to monitor the reaction in real-time to guide intervention and (iv) the advantages of nanoconfinement for enzyme cascades. A new approach for one-pot deracemisation and stereoinversion using a single electrode is thereby demonstrated. Although high enantiomeric excesses have already been achieved, 82–85.7%, the many factors at play – inherent enzyme rates, Km values and enantiomeric selectivity – mean there is considerable scope for improvement and bespoke optimisation.
The proof-of-concept can be taken forward in various ways, for example, an FNR variant specific for NAD(H) rather than NADP(H), will expand the scope. The platform is well suited to selective hydrogenation and deuteration. The emerging field of de novo enzyme production, through computational design and directed evolution,29 will expand the toolkit further; for example, the use of variants engineered to have built-in on-off “switches” to facilitate differential control.
C. F. M conceived the project, B. C. carried out all experiments, R. H. and N. J. T. provided T. ethanolicus variants, R. H. carried out chiral analysis, F. A. A., B. C., C. F. M. interpreted the data and wrote the manuscript.
This research was supported by a grant from the Biological and Biotechnological Research Council (Follow-on Fund, Grant BB/P023797/1), the EPA Cephalosporin Fund (CF 342) and by an ERC Advanced Grant (Grant number 742987).
Conflicts of interest
There are no conflicts to declare.References
- B. Siritanaratkul, C. F. Megarity, T. G. Roberts, T. O. M. Samuels, M. Winkler, J. H. Warner, T. Happe and F. A. Armstrong, Chem. Sci., 2017, 8, 4579–4586 RSC.
- C. F. Megarity, B. Siritanaratkul, R. S. Heath, L. Wan, G. Morello, S. R. FitzPatrick, R. L. Booth, A. J. Sills, A. W. Robertson, J. H. Warner, N. J. Turner and F. A. Armstrong, Angew. Chem., Int. Ed., 2019, 58, 4948–4952 CrossRef CAS PubMed.
- C. F. Megarity, B. Siritanaratkul, B. Cheng, G. Morello, L. Wan, A. J. Sills, R. S. Heath, N. J. Turner and F. A. Armstrong, ChemCatChem, 2019, 11, 5662–5670 CrossRef CAS.
- G. Morello, C. F. Megarity and F. A. Armstrong, Nat. Commun., 2021, 12, 340 CrossRef CAS PubMed.
- L. Wan, R. S. Heath, B. Siritanaratkul, C. F. Megarity, A. J. Sills, M. P. Thompson, N. J. Turner and F. A. Armstrong, Green Chem., 2019, 21, 4958–4963 RSC.
- R. A. Herold, R. Reinbold, C. F. Megarity, M. I. Abboud, C. J. Schofield and F. A. Armstrong, J. Phys. Chem. Lett., 2021, 12, 6095–6101 CrossRef CAS PubMed.
- L. Wan, C. F. Megarity, B. Siritanaratkul and F. A. Armstrong, Chem. Commun., 2018, 54, 972–975 RSC.
- G. Morello, B. Siritanaratkul, C. F. Megarity and F. A. Armstrong, ACS Catal., 2019, 9, 11255–11262 CrossRef CAS.
- L. Wan, R. S. Heath, C. F. Megarity, A. J. Sills, R. A. Herold, N. J. Turner and F. A. Armstrong, ACS Catal., 2021, 11, 6526–6533 CrossRef CAS.
- C. F. Megarity, T. R. I. Weald, R. S. Heath, N. J. Turner and F. A. Armstrong, ACS Catal., 2022, 12(15), 8811–8821, DOI:10.1021/acscatal.2c00999.
- G. W. Mellin and M. Katzenstein, N. Engl. J. Med., 1962, 267, 1184–1193 CrossRef CAS PubMed.
- M. E. Franks, G. R. Macpherson and W. D. Figg, Lancet, 2004, 363, 1802–1811 CrossRef CAS.
- M. R. Islam, J. G. Mahdi and I. D. Bowen, Drug Saf., 1997, 17, 149–165 CrossRef CAS PubMed.
- C. Magallanes-Noguera, M. M. Ferrari, M. Kurina-Sanz and A. A. Orden, J. Biotechnol., 2012, 160, 189–194 CrossRef CAS PubMed.
- B. Yuan, D. P. Debecker, X. Wu, J. Xiao, Q. Fei and N. J. Turner, ChemCatChem, 2020, 12, 6191–6195 CrossRef CAS.
- B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294–2312 CrossRef CAS PubMed.
- B. Yuan, D. P. Debecker, X. Wu, J. Xiao, Q. Fei and N. J. Turner, ChemCatChem, 2020, 12, 6191–6195 CrossRef CAS.
- J. S. DeHovitz, Y. Y. Loh, J. A. Kautzky, K. Nagao, A. J. Meichan, M. Yamauchi, D. W. MacMillan and T. K. Hyster, Science, 2020, 369, 1113–1118 CrossRef CAS PubMed.
- F. G. Mutti, A. Orthaber, J. H. Schrittwieser, J. G. de Vries, R. Pietschnig and W. Kroutil, Chem. Commun., 2010, 46, 8046–8048 RSC.
- C. V. Voss, C. C. Gruber, K. Faber, T. Knaus, P. Macheroux and W. Kroutil, J. Am. Chem. Soc., 2008, 130, 13969–13972 CrossRef CAS PubMed.
- R. S. Heath, M. Pontini, S. Hussain and N. J. Turner, ChemCatChem, 2016, 8, 117–120 CrossRef CAS.
- M. A. Cohen, J. S. Parratt and N. J. Turner, Tetrahedron: Asymmetry, 1992, 3, 1543–1546 CrossRef CAS.
- K. I. Ziegelmann-Fjeld, M. M. Musa, R. S. Phillips, J. G. Zeikus and C. Vieille, Protein Eng., Des. Sel., 2007, 20, 47–55 CrossRef CAS PubMed.
- M. M. Musa, J. M. Patel, C. M. Nealon, C. S. Kim, R. S. Phillips and I. Karume, J. Mol. Catal. B: Enzym., 2015, 115, 155–159 CrossRef CAS.
- W. Hummel, Appl. Microbiol. Biotechnol., 1990, 34, 15–19 CrossRef CAS.
- K. Niefind, J. Müller, B. Riebel, W. Hummel and D. Schomburg, J. Mol. Biol., 2003, 327, 317–328 CrossRef CAS PubMed.
- C. W. Bradshaw, W. Hummel and C. H. Wong, J. Org. Chem., 1992, 57, 1532–1536 CrossRef CAS.
- J. M. Patel, M. M. Musa, L. Rodriguez, D. A. Sutton, V. V. Popik and R. S. Phillips, Org. Biomol. Chem., 2014, 12, 5905–5910 RSC.
- S. L. Lovelock, R. Crawshaw, S. Basler, C. Levy, D. Baker, D. Hilvert and A. P. Green, Nature, 2022, 606, 49–58 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc03638j |
| ‡ Current address: School of Chemistry, University of Manchester, Manchester Institute of Biotechnology, 131 Princess Street, Manchester M1 7DN, UK. |
| This journal is © The Royal Society of Chemistry 2022 |