
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeveloping an atroposelective dynamic kinetic resolution of MRTX1719 by resolving incompatible chemical operations†
Michal M.
Achmatowicz‡
,
Cheng-yi
Chen
and
David R.
Snead‡
 *
*
Mirati Therapeutics, 3545 Cray Ct., San Diego, 92121, CA, USA. E-mail: sneadd@mirati.com
First published on 24th August 2022
Abstract
A high-yielding protocol for atropisomeric resolution was developed by rectifying incompatibilities between crystallization and epimerization via continuous processing. Application toward synthesis of MRTX1719, a densely functionalized active pharmaceutical ingredient (API), improved yield from 37% to 87%. This protocol provides a complementary means to access rotamers which challenge current asymmetric methodologies, and greatly improves sustainability by decreasing the consumption of solvent and advanced synthetic intermediates.
A high yielding protocol for obtaining enantiopure atropisomers1 is presented. Operating under a continuous flow regime overcame chemical incompatibilities to achieve a formal dynamic kinetic resolution (DKR). Atropisomers are a common subset of organocatalysts and ligands,2 and they possess intriguing properties for materials.3 However, the prominence of this structural feature in biologically active compounds is perhaps its most important attribute. A growing number of pharmaceuticals contain axial chirality.4
MRTX1719 (1a) is an emerging oncology therapy with an axis of chirality caused by a restricted rotation around the bond linking the pyrazole and benzene rings (Fig. 1).5 The barrier to rotation is only 28.9 kcal mol−1 despite the dense functionalization surrounding the biaryl bond. This results in a 1 h half-life for optically pure material at 80 °C, and complicates attempts to develop a stereoselective biaryl coupling. Even if bond-forming was stereoselective, the compound would epimerize at reaction temperatures. Moreover, axially chiral benzonitriles are known to be particularly challenging substrates.6
 | ||
| Fig. 1 MRTX1719, an atropisomeric drug candidate. | ||
Fortunately, a late-stage stereoselective crystallization was developed (Fig. 2). Optical resolutions present a straightforward protocol for producing enantiopure material, but their low yield is a serious deficiency.7 Rejection of the undesired epimer results in a loss of at least 50% of the material, and for 2a the yield was only 37%.
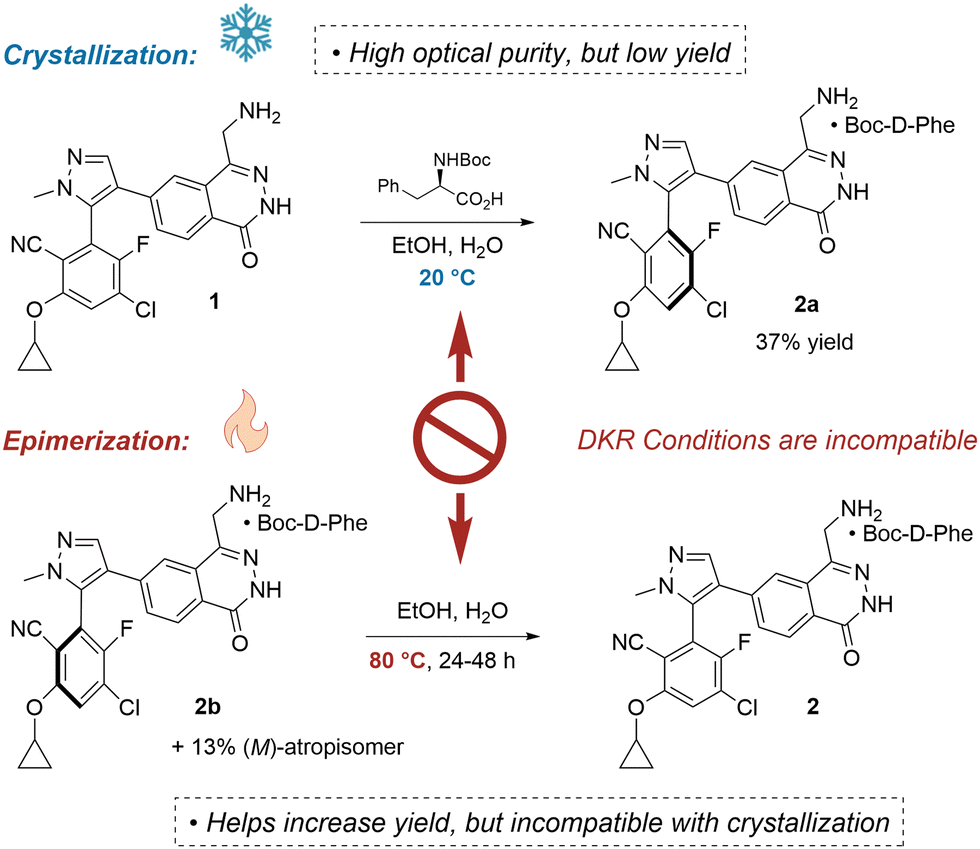 | ||
| Fig. 2 Optical resolution of 1 provides enantiopure compound, but achieving high yield via a DKR is infeasible due to conflicting requirements of the epimerization. | ||
Perhaps one might consider a dynamic kinetic resolution (DKR)8 or crystallization induced diastereomeric transformation (CIDT)9 to solve this issue. In the scenario where one stereoisomer is selectively insoluble, one could first conduct a crystallization, and then renew the population of the depleted rotamer via epimerization. Heating atropisomers can regenerate the desired stereoisomer by promoting rotation about the axis of chirality.10 Further crystallization would drive the equilibrium to completion. Unfortunately, the conditions associated with crystallization (low-temperature) and epimerization (high-temperature) are often incompatible.
Continuous flow synthesis can decouple these conflicting needs (Fig. 3). Crystallization could be conducted in a typical batch vessel. Mother liquors would then be continuously removed from the vessel by pumping through a filter. The stream containing the undesired enantiomer would be pumped through a high temperature unit to effect instant epimerization. Solution now enriched with the desired atropisomer would return to the batch vessel for further crystallization. The recycle could be conducted essentially ad infinitum to achieve a theoretical yield close to 100% (accounted for by supernatant loss). We refer to this continuous recycle with spatially discrete unit operations as a DKR mediated by simultaneous processing of antagonistic chemical events (SPACE-DKR). This strategy has very recently been applied for a select few asymmetric transformations (amine epimerization, olefin epimerization).11
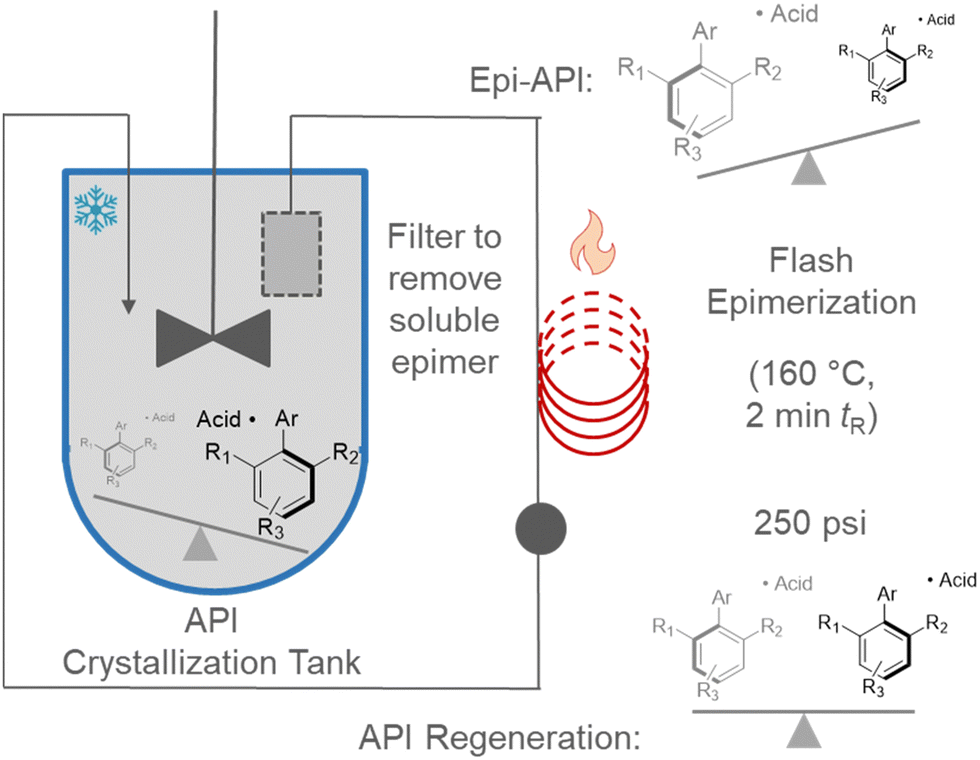 | ||
| Fig. 3 Application of continuous flow for simultaneous processing of antagonistic chemical events (SPACE). | ||
The concept was first tested with 1,1′-bi-2-naphthol (BINOL) as a model system.12 BINOL is an important ligand and chemical building block.3,13 It contains an axis of chirality with a high barrier to rotation, which renders it difficult to epimerize. Even so, an 80% overall yield was achieved by separating the crystallization and epimerization events.
After establishing proof-of-concept, attention turned to development of a DKR for the highly elaborated API, MRTX1719 (Fig. 4). There was concern whether the flash epimerization at high temperatures would be feasible given the dense functionalization of the API (primary amine, electrophilic benzene ring, cyclopropyl moiety). As such, flash epimerization was first studied in batch using microwave irradiation. Rapid heating of mother liquors showed full epimerization of substrate within 4 minutes at 160 °C with negligible loss of assay as judged by HPLC.
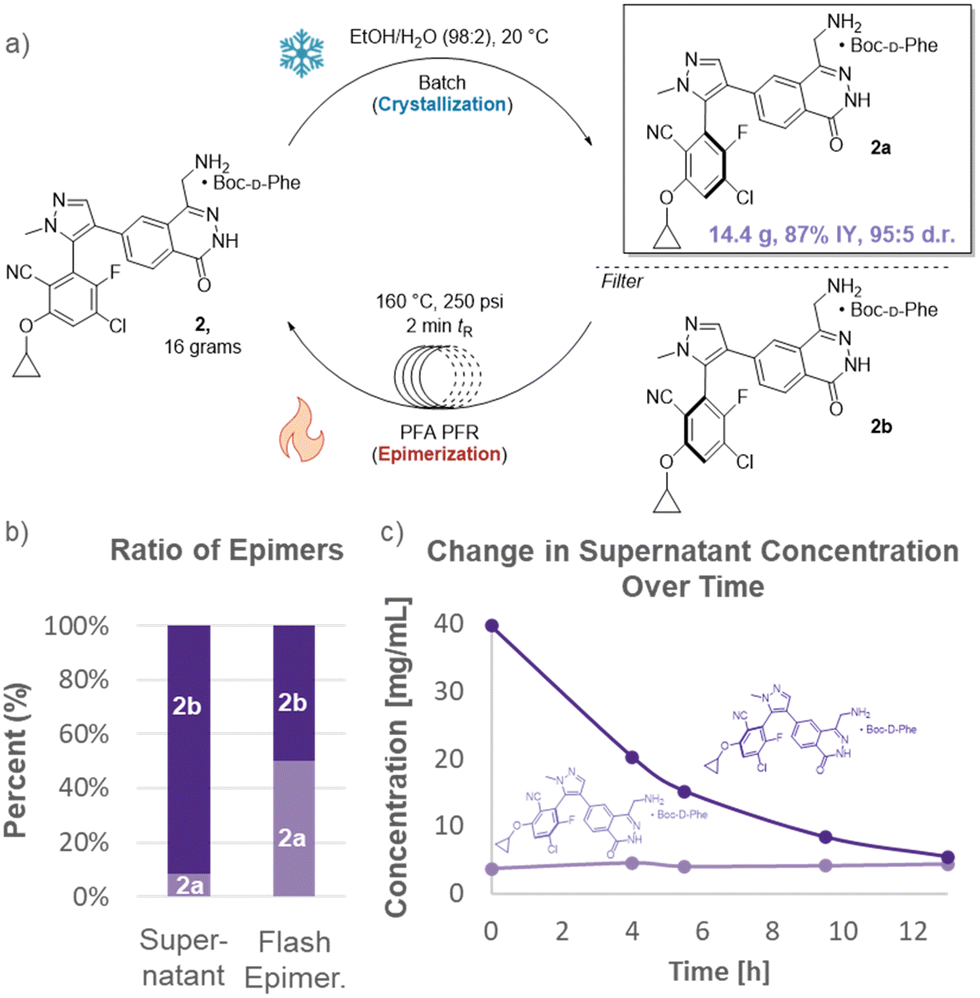 | ||
| Fig. 4 (a) SPACE-DKR of MRTX1719, improving yield to 87%. (b) Flash epimerization to enrich supernatant with 2a. (c) Depletion of undesired enantiomer, 2b, in supernatant. | ||
A combined batch-flow reactor was constructed for the DKR. Boc-protected phenylalanine was added to MRTX1719 dissolved in ethanol/water (98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio). The mixture was stirred at 20 °C overnight in a traditional batch vessel to crystallize the desired isomer. The flash epimerization was then operated by filtering the supernatant through a 20 μm filter and into a plug flow reactor (PFR) held at a temperature of 160 °C and 250 psi. After a 2 min residence time (tR), the stream exiting the PFR was fully epimerized (49.6:50.4 ratio) as it returned to the crystallization tank. The epimerization results in continual supersaturation of 2a in the batch crystallization unit until the great majority of 2b is converted to 2a and trapped via crystallization. After 14.5 h, the supernatant reached a nearly 1:1 ratio of epimers. Flash epimerization can no longer cause supersaturation of 2a, thus removing the driving force for further crystallization and yield increase. Analogous to the batch operation, residual epimer was rinsed out by slurrying the product in aqueous ethanol after the crystallization.12
:2 ratio). The mixture was stirred at 20 °C overnight in a traditional batch vessel to crystallize the desired isomer. The flash epimerization was then operated by filtering the supernatant through a 20 μm filter and into a plug flow reactor (PFR) held at a temperature of 160 °C and 250 psi. After a 2 min residence time (tR), the stream exiting the PFR was fully epimerized (49.6:50.4 ratio) as it returned to the crystallization tank. The epimerization results in continual supersaturation of 2a in the batch crystallization unit until the great majority of 2b is converted to 2a and trapped via crystallization. After 14.5 h, the supernatant reached a nearly 1:1 ratio of epimers. Flash epimerization can no longer cause supersaturation of 2a, thus removing the driving force for further crystallization and yield increase. Analogous to the batch operation, residual epimer was rinsed out by slurrying the product in aqueous ethanol after the crystallization.12
The overall yield of highly enantioenriched material increased nearly fifty percent to 87%. This greatly enhances sustainability by reducing the process mass intensity (PMI) and required input (utilization factor) of advanced synthetic intermediates by 40% (each synthon requires at least 7 synthetic steps).
SPACE-DKR can render high-yielding atroposelective syntheses a mechanical effort after a suitable resolution agent or conglomerate is identified. It is envisioned that this will provide a complementary means to asymmetric catalysis for challenging axially chiral structures. Further, the strategy will likely expand to include additional epimerizable features and more broadly impact the field of asymmetric synthesis.
The authors wish to express gratitude to Jeffrey Gustafson of SDSU and Allan Myerson of MIT for insightful conversation and guidance regarding atropisomers (J. G.) and crystallization processes (A. M.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384 CrossRef CAS PubMed.
- Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534 CrossRef CAS PubMed.
- (a) L. Pu, 1,1′-Binaphthyl-Based Chiral Materials, Imperial College Press, 2009 CrossRef; (b) A. Shockravi, A. Javadi and E. Abouzari-Lotf, RSC Adv., 2013, 3, 6717 RSC.
- (a) J. Clayden, W. J. Moran, P. J. Edwards and S. R. LaPlante, Angew. Chem., Int. Ed., 2009, 48, 6398 CrossRef CAS PubMed; (b) S. R. LaPlante, L. D. Fader, K. R. Fandrick, D. R. Fandrick, O. Hucke, R. Kemper, S. P. F. Miller and P. J. Edwards, J. Med. Chem., 2011, 54, 7005 CrossRef CAS PubMed; (c) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562 RSC; (d) P. W. Glunz, Bioorg. Med. Chem. Lett., 2018, 28, 53 CrossRef CAS PubMed.
- C. R. Smith, R. Aranda, T. P. Bobinski, D. M. Briere, A. C. Burns, J. G. Christensen, J. Clarine, L. D. Engstrom, R. J. Gunn, A. Ivetac, R. Jean-Baptiste, J. M. Ketcham, M. Kobayashi, J. Kuehler, S. Kulyk, J. D. Lawson, K. Moya, P. Olson, L. Rahbaek, N. C. Thomas, X. Wang, L. M. Waters and M. A. Marx, J. Med. Chem., 2022, 65, 1749 CrossRef CAS PubMed.
- Y. Lv, G. Luo, Q. Liu, Z. Jin, X. Zhang and Y. R. Chi, Nat. Commun., 2022, 13, 27813 Search PubMed.
- L. Derdour, E. J. Chan and D. Skliar, Processes, 2019, 7, 611 CrossRef CAS.
- R. S. Ward, Tetrahedron: Asymmetry, 1995, 6, 1475 CrossRef CAS.
- K. M. J. Brands and A. J. Davies, Chem. Rev., 2006, 106, 2711 CrossRef CAS PubMed.
- D. C. Patel, R. M. Woods, Z. S. Breitbach, A. Berhod and D. W. Armstrong, Tetrahedron: Asymmetry, 2017, 28, 1557 CrossRef CAS.
- (a) G. Valenti, P. Tinnemans, I. Baglai, W. L. Noorduin, B. Kaptein, M. Leeman, J. H. ter Horst and R. M. Kellogg, Angew. Chem., Int. Ed., 2021, 60, 5279 CrossRef CAS PubMed; (b) M. H. T. Kwan, J. Breen, M. Bowden, L. Conway, B. Crossley, M. F. Jones, R. Munday, N. P. B. Pokar, T. Screen and A. J. Blacker, J. Org. Chem., 2021, 86, 2458 CrossRef CAS PubMed; (c) J. D. Williams, P. Pöchlauer, Y. Okumura, Y. Inami and C. O. Kappe, Chem. – Eur. J., 2022, 28, e202200741 CrossRef CAS PubMed.
- See ESI† for details.
- (a) M. Kawashima and A. Hirayama, Chem. Lett., 1990, 2299 CrossRef CAS; (b) M. Kawashima and R. Hirata, Bull. Chem. Soc. Jpn., 1993, 66, 2002 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc03627d |
| ‡ M. M. A. and D. R. S. contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |