
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalysed amplification of faradaic shotgun tagging in ultrasensitive electrochemical immunoassays†
Mohamed
Sharafeldin
 ,
Robert
Hein
and
Jason J.
Davis
*
,
Robert
Hein
and
Jason J.
Davis
*
Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QZ, UK. E-mail: jason.davis@chem.ox.ac.uk
First published on 9th August 2022
Abstract
We introduce a novel electrochemical protein quantitation based on the shotgun biotin tagging of proteins prior to their interfacial immunocapture and polymeric enzyme tagging. The highly amplified faradaic signals generated from a novel ferrocene-tyramine adduct enable fg mL−1 (attomolar) levels of detection and span cross a 5 orders of magnitude dynamic range. This work supports ultrasensitive protein marker detection in a single antibody immunoassay format.
Electrochemical sensors have underpinned significant advances in disease diagnostics, environmental monitoring, and food quality control.1 Within these, performance improvements (sensitivity and/or selectivity) have been supported by several strategies such as engineering electrode surface characteristics (either by increasing surface area or conductance),2 decreasing the susceptibility of the electrode surface to interfering species (antifouling surface modification),3–5 and using amplifying labels to boost redox signals.6,7 The latter is the most commonly employed approach to increase signal/noise and to maintain control over sensor dynamic range (for example by modifying the number of enzyme tags/binding event). A diverse body of amplifying labels have been reported for electrochemical sensors where enzymes such as horseradish peroxidase, alkaline phosphatase, and glucose oxidase are commonly used.8 A growing library of inorganic enzyme mimetics have also been proposed as “more robust” alternatives to natural enzymes.9,10 The vast majority of such amplifying labels are anchored to the antigens captured on an electrode surface through a secondary labelling process mandating the use of paired antibodies or aptamer probes which is reflected by increasing assay cost and time. In contrast to the immense research directed towards developing new enzyme mimetics or polymeric enzyme labels, very little reported work has been directed towards the development of new enzyme (or mimic) substrates. Horseradish peroxidase is arguably the most commonly used enzyme for signal amplification by virtue of its low cost, high stability, good catalytic activity and substrate abundance.11 Peroxidase substrates have dominated developments in derived optical sensors but only few of these substrates have been applied to electrochemical sensors, where 3,3′,5,5′-tetramethylbenzidine (TMB) is the most commonly used.12 The HRP catalysed oxidation of TMB into oxidized TMB (where enzyme catalytic sites are recycled by solution phase hydrogen peroxide) has been employed in prior precipitate-based electrochemical sensors (with HRP labelled antibodies).4,13 This rather crude approach can be problematic and generate false negative or false positive results and high levels of inter-assay variation; both soluble and insoluble oxidation products are generated as well as some uncontrolled dimerization of the TMB radicals, some of which may be removed during washing.14 The direct peroxidase activity on hydrogen peroxide can also be used in the generation of quantifiable signatures, either directly (peroxide voltammetry) or mediated.10,15 In all of these cases enzyme (target) specific presence arises from its coupling to secondary antibodies or nucleic acid probes, necessitating more steps, more variables, a greater assay time, and substantial cost.16–18
It is well-established that HRP will catalytically activate tyramine in the presence of hydrogen peroxide generating highly reactive tyramine radicals in a process that has been applied, with dye-functionalized tyramine derivatives, to signal amplification in fluorescent imaging; the tyramine signal amplification (TSA) or catalysed reporter deposition (CARD) methods. Integral in this is the covalent coupling of generated tyramine radicals to protein tyrosine moieties.19,20 This approach has also been applied for electrochemiluminescence,21 and surface-enhanced Raman spectroscopy (SERS).22 The HRP – tyramine system has also been utilised within “standard” dual antibody sandwich electrochemical assays with additional complex further amplification e.g. the use of heavily modified nanoparticles or additional polymerisation steps.23,24
Herein, we modify our previously developed faradaic shotgun approach25 in biotin tagging targets prior to their immunocapture and facile labelling with amplifying enzymes. The single antibody system capitalizes on further amplification of electrochemical of signal using a novel HRP substrate. Enzymic turnover of ferrocene-tyramine (Fc-Ty) specifically showers electrode-confined proteins with a covalently tethered ferrocene (Fig. S1 and S2, ESI†), generating a specific faradaic signal (overcoming the drawbacks associated with the use of conventional TMB strategies where noncovalent adsorption is dominant). With streptavidin-poly(HRP) (St-poly(HRP)) (Scheme 1) this assay format is capable of exceptional levels of sensitivity (down to fg mL−1; attomolar concentration of protein) in serum. Substrate incubation times can be tailored to allow a tuning of the assay dynamic range; extending it from few pg mL−1 to tens of ng mL−1 or proteins.
 | ||
| Scheme 1 Schematic depiction of the shotgun biotinylation of antigens using biotin-LC-NHS followed by specific immune-recruitment at antibody coated electrodes. Captured targets are then incubated with streptavidin-poly(HRP), washed and incubated with ferrocene-tyramine (Fc-Ty) in the presence of hydrogen peroxide. The activated tyramine surface tethering to local tyrosine moieties of adjacent proteins generates a robust voltammetric signal that scales very sensitively with target concentration. | ||
Surface plasmon resonance (SPR) was initially employed to confirm target (α-Syn) biotinylation, the specificity of its recruitment at antibody (Ab) interfaces, and then the specificity and efficacy of St-poly(HRP) recruitment. Biotin-LC-NHS was used to shotgun biotinylate proteins at a concentration of 200 μM (≈ 20 times the approximate molar concentration of total protein in 1% human serum) introducing 3–5 biotin tags per protein marker (materials and methods, ESI†).26,27 While both biotinylated and non-biotinylated specific antigens are, of course, recruited onto the Ab-coated SPR chips, only the former shows a concentration dependent response after exposure to St-poly(HRP), confirming the specificity of HRP surface recruitment (Fig. S3, ESI†). This also confirms negligible background contributions from any unreacted biotin-LC-NHS.
We used a simple coupling reaction (see Materials and methods, ESI†) to synthesize a ferrocene-tyramine adduct (Fc-Ty)28 (Fig. S1 and S2, ESI†) and tested its susceptibility to peroxidase activity. To confirm the so generated covalent tethering of Fc-Ty onto protein-functionalized electrodes, we initially used standard HRP (diluted in 1% BSA) immobilized onto an electrode surface at different concentrations prior to incubation with Fc-Ty (8 mM in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) ethanol/water in 1 mM H2O2). Fc-Ty showed a predictably HRP concentration-dependent voltammetric signature (Fig. S4 and S5, ESI†). Square wave voltammetry (SWV) was used to assess signals due its improved sensitivity as compared to differential pulse voltammetry (DPV) (Fig. S6, ESI†). In the presence of H2O2, then, HRP catalyses the conversion of Fc-Ty into highly reactive tyramide radicals that covalently couple to local tyrosine moieties (that can be found on either HRP itself, adjacent BSA or indeed any local protein).29 For proteins which are electrode surface confined/captured, the so generated electrochemical of readout supports a very sensitive quantification.
:1 (v/v) ethanol/water in 1 mM H2O2). Fc-Ty showed a predictably HRP concentration-dependent voltammetric signature (Fig. S4 and S5, ESI†). Square wave voltammetry (SWV) was used to assess signals due its improved sensitivity as compared to differential pulse voltammetry (DPV) (Fig. S6, ESI†). In the presence of H2O2, then, HRP catalyses the conversion of Fc-Ty into highly reactive tyramide radicals that covalently couple to local tyrosine moieties (that can be found on either HRP itself, adjacent BSA or indeed any local protein).29 For proteins which are electrode surface confined/captured, the so generated electrochemical of readout supports a very sensitive quantification.
We then utilized the shotgun biotinylation of α-Syn standards (spiked into 1% human serum) for the development of an electrochemical sensor on screen printed carbon electrodes (Fig. S7, ESI†). Physisorbed anti-α-Syn antibodies (on 32 electrodes housed in a conventional 96 micro-well plate) were allowed to capture biotinylated α-Syn from the shotgun tagged samples. Captured proteins were then labelled with St-poly(HRP) and incubated for 2–5 minutes (depending on the desired dynamic range, see Materials and methods, ESI†) with Fc-Ty substrate. The assays exhibit an ultra-low detection limit (down to 25 fg mL−1) and a dynamic range spanning 5 orders of magnitude up to 400 pg mL−1 (Fig. 1A). The latter can be readily tuned simply by decreasing the incubation time of the Fc-Ty; for example, the dynamic range after 2 min incubation is between 3.2 pg mL−1 to 10 ng mL−1 α-Syn (Fig. 1B).
 | ||
| Fig. 1 Calibration of α-Syn using biotin shotgun tagging with a poly(HRP) generated covalent deposition of ferrocene after incubation with tyramine-ferrocene for (A) 5 min generating a dynamic range between 25 fg mL−1 to 400 pg mL−1 and (B) for 2 min to establish a dynamic range between 3.2 pg mL−1 and 10 ng mL−1. Error bars represent one standard deviation of measurements from 4 different electrodes. Insets are representative square wave voltammograms of the tagged interface. | ||
The electrochemical response to other common interfering proteins (which of course will also be shotgun tagged in solution) was assessed by exposing anti-α-Syn decorated electrodes to human serum albumin (HSA) (2 mg mL−1), bovine serum albumin (BSA) (2 mg mL−1), human fibrinogen (2 mg mL−1), and human Syntenin-1 (Synt-1) (400 pg mL−1). After incubation with St-poly(HRP), specific responses to α-Syn are observed with negligible (<10% of specific amperometric response to α-Syn) response to these large background levels even at the simple antibody modified interfaces used here (Fig. 2A). Assays show excellent reproducibility across different electrode arrays when repeated over a period of 5 days, with inter-array standard deviations <10% and inter-electrode variations (on the same array) <15% (Fig. 2B). An analysis of assay accuracy, as tested by spiked recovery experiments in neat human serum, demonstrates (Table S1 and Fig. S8, ESI†) high precision with recoveries between 93–106% (Fig. 3).
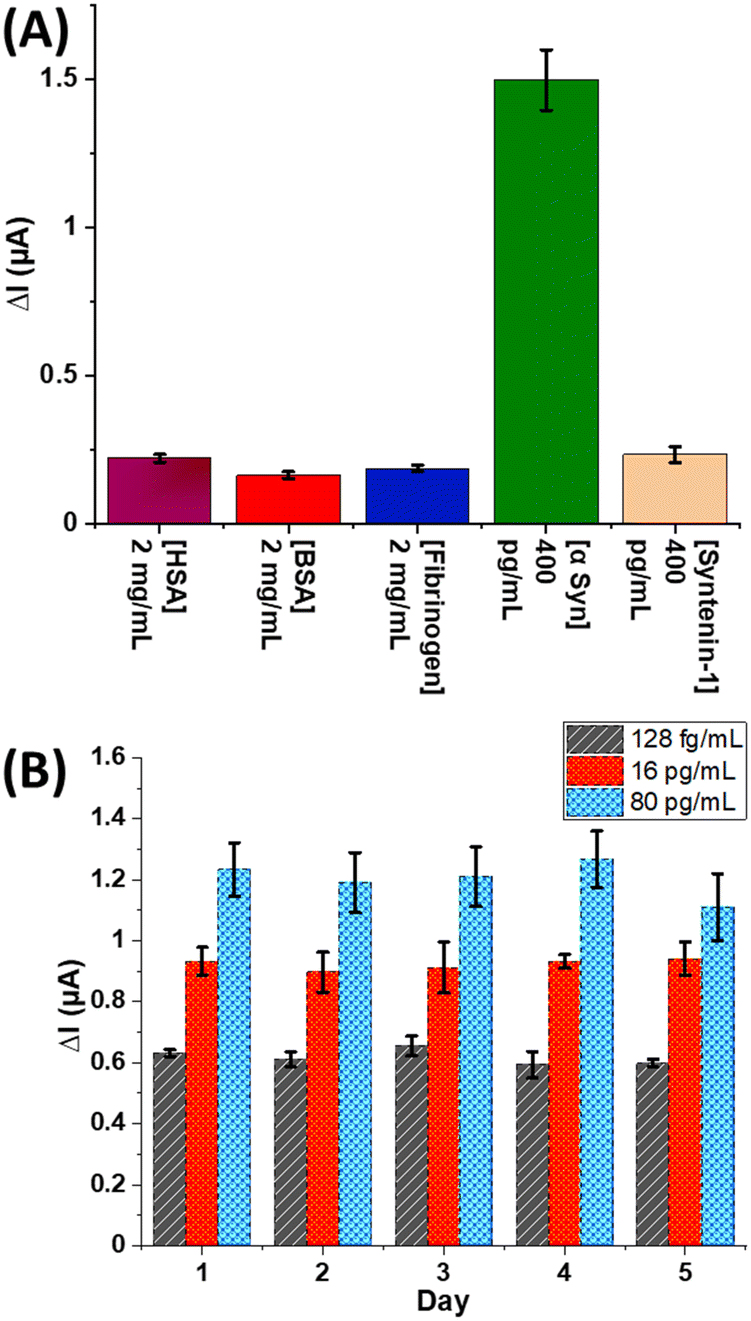 | ||
| Fig. 2 (A) Specificity study on anti-α-Syn modified electrodes against large excesses of potentially interfering proteins. Error bars represent one standard deviation from measurements across 4 different electrodes. (B) Reproducibility analyses generated by running the same concentration of α-Syn for 5 days over different electrode arrays. Each measurement represents an average across 6 individual electrodes (n = 6). | ||
 | ||
| Fig. 3 Data from analysis of spiked 1% human serum with different α-Syn concentrations. (A) Showing the correlation between spiked concentrations and found concentrations of α-Syn over a wide concentration range (between 128 fg mL−1 and 400 pg mL−1). (B) Correlation between spiked and found α-Syn concentration over a narrow concentration range (between 128 fg mL−1 and 25 pg mL−1). The regression coefficient R2 = 0.998 and slope of correlation is 1.01 indicating excellent agreement. Error bars represent standard deviations from repeats (n = 4). | ||
The sensory format developed here utilises a shotgun biotinylation and an interfacial immunocapture; the former enables the secondary tag to be a polymeric HRP. When this is coupled to a novel electrochemically active peroxidase substrate (Fc-Ty) extreme levels of assay sensitivity are accessible in assays that perform well in serum and avoid the drawbacks associated with physically adsorbed reagents (for example TMB) where nonspecific TMB adsorption can be highly problematic. The fact that these assays only use a primary antibody for selective labelled target recruitment directly reduces assay cost and time; most labelled assays require the introduction of a secondary antibody step before an additional labelling step; this requires 1–2 h and ∼20–30% of the total assay cost ($10–50/assay). The introduced configuration is easily integrated into a screen-printed 32 electrode array housed in an ELISA-compatible 96-well plate enabling high throughput sample analysis within an ELISA-like workflow. The samples and reagents can be loaded manually or using a conventional automated ELISA analyser; allowing the rapid deployment into laboratories without the need for either significant hardware or special training.
The attomolar detection limits enable the detection of proteins that are expressed at levels that few assays can access.30,31 Usefully, this approach can also be tailored to the quantification of markers at much higher (ng mL−1) levels and is readily extrapolated to any marker for which there is a well-established receptor. As tested here with spike recovery experiments in neat human serum, (ESI,† Table S1) high levels of analytical precision are possible, with recoveries between 93–106% (Fig. 3).
In conclusion, we have introduced a new sensor workflow capable of supporting protein quantitation down to few fg mL−1 concentration (attomolar levels) using a single recruiting antibody. The assay employs a common biotinylating reagent (biotin-LC-NHS) to shotgun tag samples prior to specific interfacial capture. Exemplified here with α-Syn (as a Parkinson's-relevant model target biomarker), captured targets are then coupled to St-poly(HRP) which catalyses the covalent tagging of local protein with redox addressable ferrocenes. The resulting voltammetric current reports on target analyte concentration with high specificity, selectivity and reproducibility.
Conflicts of interest
There are no conflicts to declare.References
- M. Sharafeldin and J. J. Davis, Anal. Chem., 2021, 93, 184–197 CrossRef CAS PubMed.
- S. A. Lim and M. U. Ahmed, RSC Adv., 2016, 6, 24995–25014 RSC.
- C. Jiang, G. Wang, R. Hein, N. Liu, X. Luo and J. J. Davis, Chem. Rev., 2020, 120, 3852–3889 CrossRef CAS PubMed.
- S. S. Timilsina, N. Durr, M. Yafia, H. Sallum, P. Jolly and D. E. Ingber, Adv. Healthcare Mater., 2021, 2102244 Search PubMed.
- P.-H. Lin and B.-R. Li, Analyst, 2020, 145, 1110–1120 RSC.
- Y. Liu, Y. Liu, L. Qiao, Y. Liu and B. Liu, Curr. Opin. Electrochem., 2018, 12, 5–12 CrossRef CAS.
- V. Ruiz-Valdepeñas Montiel, E. Povedano, E. Vargas, R. M. Torrente-Rodríguez, M. Pedrero, A. J. Reviejo, S. Campuzano and J. M. Pingarrón, ACS Sens., 2018, 3, 211–221 CrossRef PubMed.
- K. Thapa, W. Liu and R. Wang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2022, 14, e1765 CAS.
- K. Białas, D. Moschou, F. Marken and P. Estrela, Microchim. Acta, 2022, 189, 172 CrossRef PubMed.
- M. Sharafeldin, G. W. Bishop, S. Bhakta, A. El-Sawy, S. L. Suib and J. F. Rusling, Biosens. Bioelectron., 2017, 91, 359–366 CrossRef CAS PubMed.
- Y. Xianyu, Y. Chen and X. Jiang, Anal. Chem., 2015, 87, 10688–10692 CrossRef CAS PubMed.
- E. S. Bos, A. A. van der Doelen, N. v Rooy and A. H. W. M. Schuurs, J. Immunoassay, 1981, 2, 187–204 CrossRef CAS PubMed.
- P. Fanjul-Bolado, M. B. González-García and A. Costa-García, Anal. Bioanal. Chem., 2005, 382, 297–302 CrossRef CAS PubMed.
- U. Zupančič, P. Jolly, P. Estrela, D. Moschou and D. E. Ingber, Adv. Funct. Mater., 2021, 31, 2010638 CrossRef.
- A. L. Jones, L. Dhanapala, T. A. Baldo, M. Sharafeldin, C. E. Krause, M. Shen, S. Moghaddam, R. C. Faria, D. K. Dey, R. W. Watson, R. Andrawis, N. H. Lee and J. F. Rusling, Anal. Chem., 2021, 93, 1059–1067 CrossRef CAS PubMed.
- B. Cai and C. J. Krusemark, Angew. Chem., Int. Ed., 2022, 61, e202113515 CAS.
- D. M. O'Hara, V. Theobald, A. C. Egan, J. Usansky, M. Krishna, J. TerWee, M. Maia, F. P. Spriggs, J. Kenney, A. Safavi and J. Keefe, AAPS J., 2012, 14, 316–328 CrossRef PubMed.
- M. Spengler, M. Adler and C. M. Niemeyer, Analyst, 2015, 140, 6175–6194 RSC.
- M. R. Clutter, G. C. Heffner, P. O. Krutzik, K. L. Sachen and G. P. Nolan, Cytometry, Part A, 2010, 77, 1020–1031 CrossRef PubMed.
- L. Faget and T. S. Hnasko, in ELISA: Methods and Protocols, ed. R. Hnasko, Springer New York, New York, NY, 2015, pp. 161–172 Search PubMed.
- J.-T. Cao, L.-Z. Zhao, Y.-Z. Fu, X.-M. Liu, S.-W. Ren and Y.-M. Liu, Sens. Actuators, B, 2021, 331, 129427 CrossRef CAS.
- C. Fu, S. Jin, W. Shi, J. Oh, H. Cao and Y. M. Jung, Anal. Chem., 2018, 90, 13159–13162 CrossRef CAS PubMed.
- L. Hou, Y. Tang, M. Xu, Z. Gao and D. Tang, Anal. Chem., 2014, 86, 8352–8358 CrossRef CAS PubMed.
- L. Yuan, L. Xu and S. Liu, Anal. Chem., 2012, 84, 10737–10744 CrossRef CAS PubMed.
- M. Sharafeldin, F. Fleschhut, T. James and J. J. Davis, Anal. Chem., 2022, 94, 2375–2382 CrossRef CAS PubMed.
- J. A. Bradburne, P. Godfrey, J. H. Choi and J. N. Mathis, Appl. Environ. Microbiol., 1993, 59, 663–668 CrossRef CAS PubMed.
- M. Leeman, J. Choi, S. Hansson, M. U. Storm and L. Nilsson, Anal. Bioanal. Chem., 2018, 410, 4867–4873 CrossRef CAS PubMed.
- A. H. N. Hopman, F. C. S. Ramaekers and E. J. M. Speel, J. Histochem. Cytochem., 1998, 46, 771–777 CrossRef CAS PubMed.
- M. Kurisawa, F. Lee, L.-S. Wang and J. E. Chung, J. Mater. Chem., 2010, 20, 5371–5375 RSC.
- M. Norman, T. Gilboa and D. R. Walt, Clin. Chem., 2022, 68, 431–440 CrossRef PubMed.
- C. Wu, T. J. Dougan and D. R. Walt, ACS Nano, 2022, 16, 1025–1035 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc03509j |
| This journal is © The Royal Society of Chemistry 2022 |