
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-mild synthesis of nanometric metal chalcogenides using organyl chalcogenide precursors
Shashank
Mishra
 *
*
Université Claude Bernard Lyon 1, CNRS, UMR 5256, Institut de Recherches sur la Catalyse et l’Environnement de Lyon (IRCELYON), 2 Avenue Albert Einstein, 69626 Villeurbanne, France. E-mail: shashank.mishra@ircelyon.univ-lyon1.fr; Fax: +33-472445399; Tel: +33-472445322
First published on 5th August 2022
Abstract
Bis(trialkylsilyl) monochalcogenides and diorganyl dichalcogenides, (R3Si)2E and R2E2 (E = S, Se or Te and R = alkyl, aryl or allyl group), have emerged in the past decade as excellent reagents for the synthesis of metal chalcogenide nanoparticles (NPs) and clusters owing to their ability to transfer the chalcogenide anion (E2−) under ultra-mild conditions and versatility in reacting even with non-conventional metal reagents or being employed in a variety of synthetic methods. In comparison, the related non-silylated diorganyl monochalcogenides R2E have received attention only recently for the solution phase synthesis of metal chalcogenide NPs. In spite of sharing many similarities, these three families of organyl chalcogenides are different in their coordination ability and decomposition behavior, and therefore in reactivities towards metal reagents. This feature article provides a concise overview on the use of these three families as synthons for the ultralow-temperature synthesis of metal chalcogenide nanomaterials, deliberating their different decomposition mechanisms and critically assessing their advantages for certain applications. More specifically, it discusses their usefulness in (i) affording molecular precursors with different kinetic and thermal stabilities, (ii) isolating reactive intermediates for comprehending the mechanism of molecule-to-nanoparticle transformation and, therefore, achieving fine control over the synthesis, (iii) stabilizing isolable metastable or difficult-to-achieve phases, and (iv) yielding complex ternary nanoparticles with controlled stoichiometry or composites with sensitive materials without modifying the characteristics of the latter. Besides providing a perspective on the low-temperature synthesis of nanomaterials, this overview is expected to assist further progress, particularly in the field of R2E, leading to interesting materials including metastable ones for new applications.
 Shashank Mishra | Shashank Mishra is Associate Professor of Chemistry at the Université Claude Bernard Lyon 1, France. His research interests are in the intersection of molecular and materials chemistry and mainly concerned with the design of molecular precursors and understanding their transformations to metal chalcogenide, metal oxide and metal fluoride nanomaterials via solution or vapor phase routes for applications in optics, energy, catalysis and related fields. He has also made contributions in the field of halometallate-based perovskite-inspired materials for photovoltaics. About 85 research articles, 6 reviews, 2 book chapters, and over 35 invited talks in international conferences summarize his research activities in the above field. Being a recipient of a couple of young scientist awards in the past, he was recently named an Emerging Investigator 2022 by the ACS journal Crystal Growth & Design. |
1. Introduction
Metal chalcogenide nanomaterials have been attracting world-wide attention as semiconducting materials with size-dependent and, hence, tunable electronic and optical properties (e.g., band gap energy, carrier density, and dielectric response), which make them very attractive for several applications such as thermoelectricity, photovoltaics, photocatalysis, displays, biomedical imaging, etc.1–16 The chemical and electronic properties of these functional materials are often governed by the synthetic methods employed. An ideal synthetic method must afford these materials in a reproducible manner and with a very high level of control over their size, shape, morphology and size dispersion.1–5,15,16 Since the ‘hot injection’ method reported in 1993 by Murray et al. for the controlled synthesis of monodisperse nanoparticles (NPs) of CdE (E = S, Se, Te),17 several solution-phase methods involving either single source precursors or separate metal and chalcogenide reagents have been developed for the synthesis of colloidal metal chalcogenide NPs. These employ different chalcogenide ligands/reagents such as tertiary phosphine chalcogenides, chalcogenoureas, chalcogenolates, chalcogenocarbamates, xanthates, dichalcogenoimidodiphosphinates, or sulfur and selenium dissolved in octadecene or amines.18–20 Although these precursors have many advantages such as easy availability and high solubility in organic solvents, a majority of them require high temperature, which favors the formation of thermodynamically stable species only. Lowering the synthesis temperature is a highly desirable step to use these materials in technologically advanced areas such as energy conversion and storage, catalysis, environment, biomedicine and electronics.Unlike metal oxides,21–24 the low-temperature synthesis of functional metal chalcogenide nanomaterials is yet to realize its potential, and an important area in this direction is the exploration and identification of the reagents and precursors that are reactive at low temperature.25,26 Other reasons behind exploring alternative chalcogenide reagents include cost, ease of handling, toxicity, purity, and the ability to tune the nucleation and growth kinetics. In this regard, bis(trialkylsilyl) monochalcogenides (R3Si)2E and diorganyl dichalcogenides R2E2 (where E = S, Se or Te, and R = an alkyl, aryl or allyl group) have emerged as excellent entry points for the synthesis of metal chalcogenide nanomaterials and clusters, owing to (i) their ability to transfer the chalcogenide anion (E2−) under ultra-mild conditions, (ii) easy tunability of their reactivity and properties by varying the organyl group R, (iii) facile removal of the byproducts, (iv) commercial availability, and (v) high solubility in common organic solvents.25–31 These versatile reagents not only are applicable in a variety of synthetic methods (co-precipitation, thermal or microwave decomposition, ionic liquid media, microemulsions, chemical vapor deposition, etc.), but can also react in a facile manner even with non-conventional metal reagents (metal oxides, metal chalcogenides, metallic nanoparticles, etc.) to yield metal chalcogenide NPs and nanoclusters. Since these reagents generate metal chalcogenide NPs under mild conditions, they can generate metastable and previously unknown crystalline phases for certain materials and can even be exploited for the synthesis of composites with sensitive materials without modifying their structural and morphological characteristics.
The related non-silylated diorganyl monochalcogenides R2E, which exhibit many similarities to the above two families of organyl chalcogenides, have received much less attention for solution-phase low temperature synthesis of metal chalcogenide nanomaterials. While as a class R2E does share many of the advantages listed above for (R3Si)2E and R2E2, it also differs in many features. As compared to (R3Si)2E, which often forms a thermally unstable R3SiE-M bond due to the excellent leaving property of the silylated group, the neutral R2E forms a stronger and stable bond with the metal centre, making them usually less efficient in transferring the chalcogen to metal centers and leading to the formation of stable metal organyl chalcogenide adducts.32,33 This restricts their use mainly to the elaboration of thin films by the chemical vapor deposition technique, either as a reagent (in dual source CVD)34–37 or as single source precursors (after complexation with metals),38–41 requiring usually high temperature. On the other hand, R2E is usually more reactive than R2E2, which has an additional E–E bond to be ruptured during the decomposition. These features make the reactivity of R2E somewhat unique, which, simply by varying R and E, can be modulated between highly reactive (R3Si)2E and somewhat less reactive R2E2. This may have implications in the potential use of R2E in many applications including in the synthesis of new metastable phases with unique properties and applications, as shown in the case of R2E2.42 Despite these unique features and high potential, the use of R2E in the low-temperature solution-phase synthesis of metal chalcogenide nanomaterials has received much less attention. Exploiting the facile decomposition mechanism in some of the non-silylated diorganyl monochalcogenides R2E, a recent study from our laboratory has shown (i) the synthesis of metal precursors that decompose at very low temperature to generate metal chalcogenide nanomaterials,43 and (ii) the direct synthesis of metal chalcogenide NPs at room temperature.44–47 In the latter case, the isolation and characterization of the reactive molecular intermediates has helped in understanding the molecule-to-nanoparticle transformation better towards the development of synthetic methods with greater control.
In spite of the great utility of these organyl chalcogenides in the solution phase synthesis of metal chalcogenide nanomaterials under ultra-mild conditions, no inclusive effort has been made so far to review the subject matter. While the use of bis(trialkylsilyl) monochalcogenides (R3Si)2E in generating metal chalcogenide molecular clusters has been reviewed,28–31 their use in the synthesis of nanomaterials only finds a somewhat limited mention in a recent book chapter.48 The use of R2E2 as synthons for semiconducting nanoparticles was reviewed in 2015,26 but there have been some important developments in the field since then, particularly in the synthesis of metastable phases.49–51 This review highlights and contextualizes the emerging work on the use of organyl chalcogenides as interesting chalcogenide reagents for the mild synthesis of metal chalcogenide nanomaterials in the solution phase. After a brief introduction, the section on non-silylated organyl monochalcogenides R2E focuses on their divergent reactivity to afford either nanoparticles or molecular complexes with different kinetic and thermal stability and underlines the importance of identifying the reactive intermediates in comprehending what happens at the interface when molecules are transformed into nanomaterials and, therefore, achieving fine control over the synthesis. The advantages of mild and controlled preparation of these NPs are discussed in terms of ligand-free synthesis of metal chalcogenide–TiO2 nanocomposites for photocatalytic applications. It is followed by a subsection on the closely related precursors (R2M)2E and (R′2M)ER (M = Sb or Bi), which can be considered as derived from R2E by replacing both or one R group(s) with the R′2M moiety (M = Sb, Bi), for binary M2E3 or ternary (SbxBi1−x)2E3 nanomaterials.52 The next section on bis(trialkylsilyl) monochalcogenides (R3Si)2E describes in detail the use of different methods and the influence of diverse reaction conditions on the synthesis of different types of metal chalcogenide nanomaterials including complex ternary nanocrystals with controlled stoichiometry. The final section on R2E2 compares its reactivity with R2E and summarizes recent important advances in its use for the isolation of new metastable phases. In the end, it identifies and signifies the areas for future research in the Conclusions and looking ahead section. I hope that this article will not only provide a reference text for a wider community working on metal chalcogenide nanomaterials, including a general perspective on the low-temperature synthesis of nanomaterials, but also initiate a future progression, particularly on non-silylated R2E, to enhance further the overall understanding of molecule-to-nanoparticle transformation and contribute to the synthesis of interesting metal chalcogenide materials for new high-tech applications.
2. Non-silylated diorganyl monochalcogenides R2E: distinctive reactivity towards metal reagents
As mentioned in the Introduction section, the utilization of nonsilylated diorganyl monochalcogenides R2E has been mainly restricted to the elaboration of thin films in the chemical vapor deposition technique which requires high temperature.34–41 Although generally less reactive in comparison to silylated organyl chalcogenides (R3Si)2E, the reactivity patterns and properties of non-silylated analogues R2E can be modulated in a subtle manner by varying systematically the organyl group R. For instance, the reactions of FeCl2 with diallyl sulfide (allyl)2S, dibenzyl sulfide Bz2S and di-tert-butyl sulfide tBu2S in oleylamine at 220 °C yielded different morphologies of pyrrhotite Fe7S8 NPs, whereas di-phenyl sulfide Ph2S under similar conditions did not react with FeCl2.27 Some of these organyl chalcogenides R2E, particularly those containing di-tertiary-butyl groups tBu2E (E = S, Se, Te), have been shown to undergo facile decomposition at low temperature, although their exact decomposition mechanism has been a subject of debate.53,54 The possible initial steps in the decomposition include (i) C–E bond cleavage to give tBu˙ and tBuE˙ radicals and (ii) β-H elimination to give isobutene and tertiary-butyl chalcogenol (Scheme 1). While it was proposed using deuterium labelled experiments that tBu2Se undergoes decomposition via initial Se–C bond cleavage,53 recently it was shown using ab initio calculations that the non-radical β-H elimination pathway could almost fully account for the decomposition of tBu2S, even though radical reactions do have an effect on the product distribution.54 This difference in the decomposition mechanism of tBu2Se and tBu2S may be traced to a slightly different strengthening of the C–S and C–Se bonds, the former being thermodynamically more stable. This facile decomposition in tBu2E (E = S, Se, Te) and related reagents can be exploited as a strategy to either prepare single source precursors with low thermal decomposition temperature or get metal chalcogenide NPs directly at room temperature, as described below.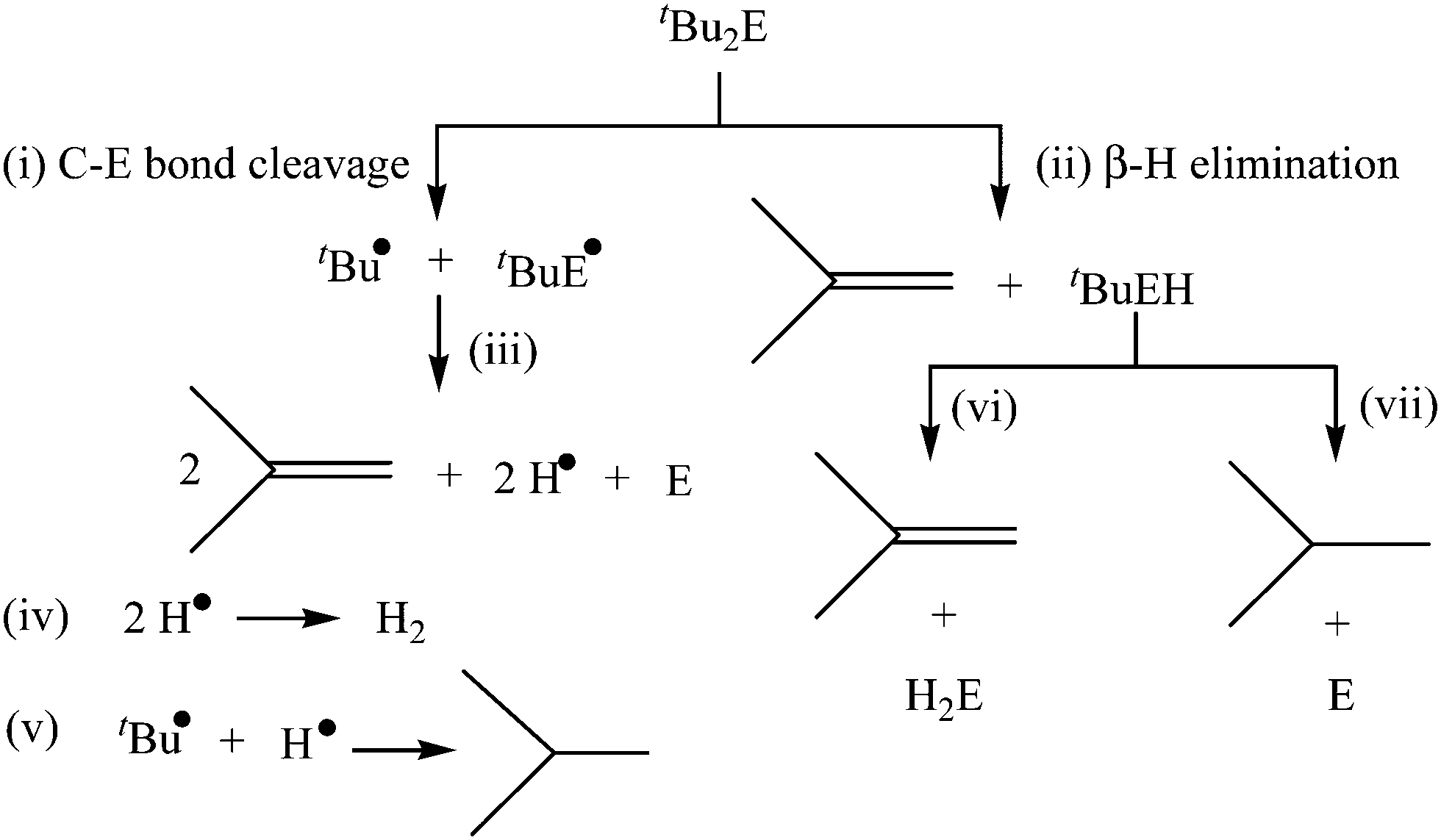 | ||
| Scheme 1 The possible decomposition pathways of tBu2E (E = S, Se). | ||
2.1. Stable molecular complexes with low thermal decomposition temperature
The high coordinating ability of organyl chalcogenides to afford molecular complexes is well-known.32,33 However, the application of these complexes as single source precursors is relatively new and mostly confined to vapor phase synthesis which requires high temperature.38–41 Employing well-characterized single-source precursors (SSPs) with well-defined composition not only overcomes the problem of controlling the stoichiometry of the constituent elements in the metal chalcogenides and paves the way for their low temperature and sustainable syntheses (therefore reducing the fabrication cost), but also provides numerous opportunities for the design and improvement in the properties of these materials.55–57 As metal chalcogenide nanostructures and thin films are the core components of high-performance devices, the low temperature solution phase fabrication of these materials offers a unique opportunity for their incorporation in relevant areas like electronics, energy conversion and storage, environmental science, catalysis and biomedicine. As mentioned previously, the kinetic and thermal stability of these molecular complexes [MXy(R2E)z]n can vary depending on the nature of the metal (M), ancillary ligand (X) or substituent R and chalcogen atom E in the organyl chalcogenides. Using R2E with alkyl groups that have a facile decomposition mechanism via β-hydrogen elimination, several molecular complexes with low thermal stability have been synthesized. For instance, Reid et al. have synthesized many metal halide complexes with the di-n-butyl chalcogenide ligand [MCl4(nBu2E)2] (M = Ti, Sn, Mo; E = S or Se), the thermogravimetric analyses (TGA) of which showed their low thermal decomposition (typically below 250 °C).58–60 The C–Se bond is slightly weaker than the C–S bond and, therefore, the nBu2Se analogues are expectedly decomposed at slightly lower temperature than those with the nBu2S ligand. These precursors were employed in MOCVD to deposit metal chalcogenide thin films. On substituting the nBu group with tBu, the decomposition can further be accelerated, thus lowering the decomposition temperature further. Fig. 1 shows the TGA curves of some homo- and heterometallic complexes of the coinage metals with the tBu2S ligand, which exhibit one- or two-step decomposition at low temperature (typically between 110 and 140 °C) with residues that correspond to metal sulfides as the end products. While homometallic copper and silver precursors [Cu2(TFA)4(tBu2S)2] and [Ag4(TFA)4(tBu2S)4] (where TFA = trifluoroacetate) are decomposed to give Cu9S5 and Ag2S, respectively,43 the heterometallic [Ag2Cu(TFA)4(tBu2S)4] decomposes in a single step at 120 °C to give a mixture of Ag3CuS2 and Ag2S.46 In contrast, organyl chalcogenides which do not have an alternative decomposition mechanism (e.g., Me2E) afford metal complexes that are kinetically and thermally stable.44,46,61 For example, the homometallic [Ag5(TFA)5(Me2Se)4] and heterometallic [Ag2Cu(TFA)4(Me2Se)4] complexes require much high temperature (∼300 °C) to decompose to yield Ag2Se and a mixture of AgCuSe + Ag2Se, respectively.44,46 | ||
| Fig. 1 Homo- and heterometallic coinage metal complexes with tBu2S as a precursor with ultra-low decomposition temperature. Adapted with permission from ref. 43 and 46. Copyright (2021) Wiley-VCH; (2020) American Chemical Society. | ||
2.2. Isolation of reactive molecular intermediates during the course of formation of metal chalcogenide nanoparticles
In the bottom-up approach of materials synthesis, identification of intermediate species operating at the intersection of molecular and nanometric levels is a key aspect in understanding the mechanism of molecule-to-nanoparticle formation and therefore achieving fine control over the synthetic conditions to prepare materials with controlled composition and properties. However, it is challenging to isolate and characterize these intermediates unambiguously because they are usually highly reactive in nature. Recently, we have found success in using R2E of suitable reactivity to isolate and characterize reactive molecular intermediates during the course of formation of binary and ternary coinage metal selenide NPs.44–46 Specifically, by reacting tBu2Se with Cu(TFA)2 or/and Ag(TFA) (where TFA = trifluoroacetate) in a variety of solvents (diethyl ether, tetrahydrofuran or toluene), we reported direct synthesis of binary and ternary coinage metal selenide NPs Cu2−xSe, Ag2Se and AgCuSe NPs at room temperature. We succeeded in isolating and characterizing by the single crystal X-ray technique the highly reactive intermediates [Cu2(TFA)2(tBu2Se)3], [Ag(TFA)(tBu2Se)2] and [Ag2Cu(TFA)4(tBu2Se)4] during the course of these reactions, which provided unambiguous proof that Cu2−xSe, Ag2Se and AgCuSe NPs are formed via the above intermediates, respectively (Fig. 2). These isolated intermediates are unstable as they turn black within a few days even at low temperature and in an inert atmosphere, apparently due to their transformation into metal selenides. The thermal and kinetic instability of these intermediates was further confirmed by thermogravimetric (TGA) and density functional theory (DFT) calculations, respectively. For example, the TGA of the heterometallic intermediate [Ag2Cu(TFA)4(tBu2Se)4] shows its decomposition below 100 °C with a residual mass that is consistent with the formation of 1 eq. AgCuSe and 0.5 eq. Ag2Se as the end product, whereas a HOMO–LUMO gap of 0.25 eV was calculated from the DFT calculations.46 The fact that this intermediate is transformed to a mixture of AgCuSe and Ag2Se when left in air for a few hours underlines its high reactivity. In comparison, the tBu2S analogue [Ag2Cu(TFA)4(tBu2S)4] shows a better kinetic stability and can be kept intact at room temperature for several weeks. The higher stability of this analogue is attributed to a stronger C–S bond (as compared to the C–Se bond). Besides being a facile source of Se2−, the coordinated tBu2Se ligand also plays the role of a reducing reagent to get the desirable +1 oxidation state of copper during the transformation of [Cu2(TFA)2(tBu2Se)3] and [Ag2Cu(TFA)4(tBu2Se)4] to Cu2−xSe and CuAgSe + Cu2−xSe NPs, respectively. In comparison to intermediates involving Cu(II) species, the transformation of [Ag(TFA)(tBu2Se)2] to Ag2Se NPs required no metal reduction step and, therefore, the reaction was much faster (5–30 min), more reproducible and afforded a better yield (>95%).44 The binary Cu2−xSe and Ag2Se NPs on further reaction with tBu2Se at RT gave compositionally tailored ternary metal chalcogenide CuAgSe NPs.47 The above ultra-mild synthesis of binary and ternary NPs using the tBu2Se ligand was further exploited to generate composites with TiO2 (P25) for photocatalysis without compromising the structural and morphological characteristics of TiO2 and without having any organics around NPs. As a result, the obtained metal–chalcogenide–TiO2 nanocomposites acted as better photocatalysts than the commercially available benchmark TiO2 (P25) for formic acid degradation under UV irradiation.44,45,47 Some selected examples of metal chalcogenide nanoparticles synthesized under mild conditions using non-silylated organyl chalcogenides are summarized in Table 1. | ||
| Fig. 2 Isolation and structural characterization of reactive intermediates during the syntheses of Cu2−xSe, Ag2Se and AgCuSe NPs from the reactions of tBu2Se with coinage metal trifluoroacetates. Adapted with permission from ref. 44–46. Copyright (2016) Wiley-VCH; (2018) The Royal Society of Chemistry; and (2020) American Chemical Society. | ||
| Reagents/precursors | Materials | Synthesis (temp., solvent, capping ligand, etc.) | Ref. | Comments |
|---|---|---|---|---|
| [Cu2(TFA)4(tBu2S)2] | ODT-Protected Cu9S5 NPs | 145 °C, xylene, ODT | 43 | Precursor with one-step decomposition (single DTG peak at 110 °C) |
| Cu(TFA)2 + tBu2Se | Cu2−xSe NPs | 110 °C, toluene | 45 | Reaction proceeds via a reactive intermediate [Cu2(TFA)2(tBu2Se)3] |
| [Ag4(TFA)4(tBu2S)4] | ODT-protected Ag2S NPs | 145 °C, xylene, ODT | 43 | Precursor decomposes at as low temperature as 60 °C |
| Ag(TFA) + tBu2Se | Ag2S NPs | RT, Et2O | 44 | A reactive intermediate [Ag(TFA)(tBu2Se)2] was isolated during the reaction and characterized structurally |
| FeCl2 + R2S (R = tBu, allyl, Bz, Ph) | Pyrrhotite Fe7S8 NPs | 220 °C, OLA | 27 | While tBu2S, (allyl)2S, and (benzyl)2S yielded the pyrrhotite phase (Fe7S8) with different morphologies, Ph2S did not react under identical conditions |
| Cu2−xSe NPs + Ag(TFA) + tBu2Se | Uncapped CuAgSe NPs | RT, THF | 47 | Ultra-mild synthesis allows to combine CuAgSe with TiO2 without modifying the characteristics of TiO2 |
| [CuAg2(TFA)4(tBu2S)4] | Ag3CuS2 NPs | 110 °C, toluene | 46 | Precursor with one-step decomposition (single DTG peak at 120 °C) |
| Ag(TFA) + Cu(TFA)2 + tBu2Se | AgCuSe NPs | RT, no solvent or capping ligand | 46 | A highly reactive intermediate [CuAg2(TFA)4(tBu2Se)4] was isolated during the reaction and characterized |
2.3. Related (R′2M)2E and (R′2M)2ER (M = Sb, Bi) as precursors for Group 15 metal chalcogenides
The related (R′2M)2E and (R′2M)ER, which can be considered as derived from R2E by replacing both or one R group(s) with the R′2M moiety (M = Sb, Bi), have recently been employed as single source precursors under a variety of mild conditions to get binary M2E3 and ternary Sb2(S1−xSex)3 or BixSb1−xE3 materials.52 Although it is known in the literature for quite some time that the related complexes of the type (RM)2E (where M = Zn, Cd, Hg; E = S, Se, Te; R = Me, Ph) decompose readily to give ME and R2M,62 it is only recently that Schulz et al. have carried out a systematic study on (R′2M)2E for group 15 binary and ternary metal chalcogenide nanomaterials.63–68 For instance, they used (Et2Sb)2E (E = S, Se, Te) as a SSP for highly crystalline Sb2E3 NPs in different organic solvents in the presence of poly(1-vinylpyrrolidone)graft-1-hexadecene (PVP*) as a capping agent at temperatures below 170 °C.63,64 While (Et2Sb)2Te decomposes upon heating at 140 °C in a straightforward manner to produce highly stoichiometric Sb2Te3 and Et3Sb, the decomposition pathways of (Et2Sb)2E (E = S, Se) are more complex and produce Sb2E3, SbEt3 and Et2E. This leads to formation of slightly Sb-rich Sb2S3 and Sb2Se3 materials. Depending upon the nature of chalcogenide atoms, the morphology of the materials also differed significantly. Thus, Sb2S3 and Sb2Se3 were obtained as nanobundles and nanowires, respectively (Fig. 3a), whereas Sb2Te3 formed hexagonal, almost monodisperse, crystalline nanoplates of roughly 400 nm diameter and 35 nm thickness (Fig. 3b). The above difference in the decomposition pathways of (Et2Sb)2E is further manifested during their co-decomposition to get possibly ternary phases. While excellent compatibility in the decomposition behaviour and morphology of (Et2Sb)2S and (Et2Sb)2Se led to the formation of the ternary Sb2(S, Se)3 phase without any impurities on their simultaneous decomposition at 170 °C, co-decomposition of (Et2Sb)2S and (Et2Sb)2Te under similar conditions yielded biphasic, heterocomposite materials containing crystalline Sb2S3 nanobundles and Sb2Te3 nanoplates (Fig. 3c).64 In contrast to the behavior of (Et2Sb)2E, the thermolysis of the analogous bismuth precursor (Et2Bi)2Te in 1,3-diisopropylbenzene (DIPB) led to the formation of Bi4Te3 at 80 °C (Fig. 3d) and mixtures of Bi4Te3 and elemental Bi at higher temperatures, most probably due to the comparably low Te–Bi binding energy and complex decomposition mechanism involving radicals.65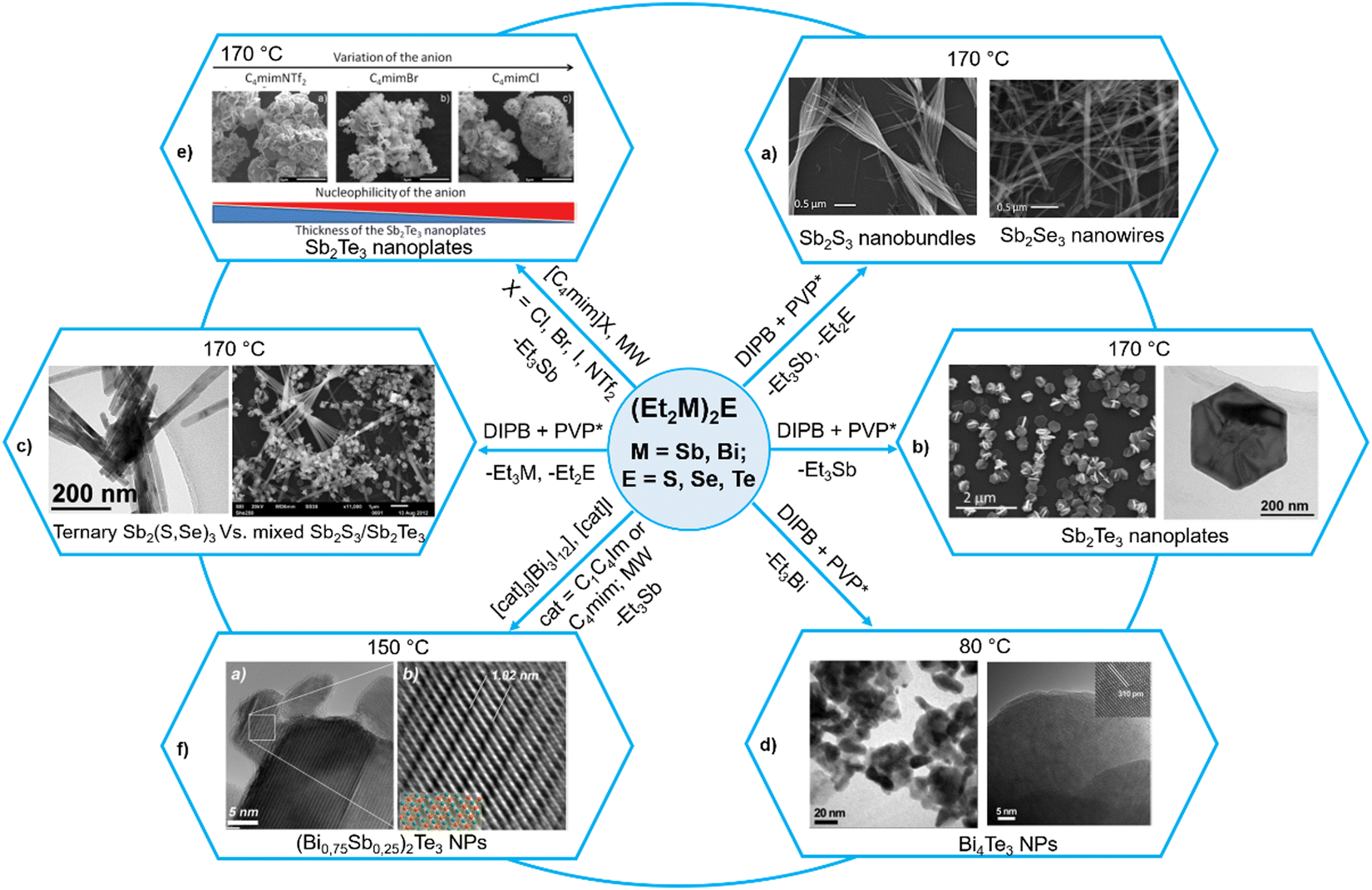 | ||
| Fig. 3 Diversity of (Et2M)2E (M = Sb, Bi; E = S, Se, Te) precursors to get binary M2E3 and ternary Sb2(S1−xSex)3 or (BixSb1−x)2Te3 nanoparticles. Adapted with permission from ref. 63–65, 67 and 68. Copyright (2015, 2021) Wiley-VCH; (2015, 2017) The Royal Society of Chemistry; and (2012) American Chemical Society. | ||
The quality of the above materials could further be improved by microwave-assisted decomposition of the above precursors in ionic liquids.66,67 Thus, microwave-assisted decomposition of (Et2Sb)2Te in [C4mim]Br (C4mim = 1-butyl-3-methyl-imidazolium) produced phase pure Sb2Te3 NPs with improved thermoelectric properties.65 The authors then studied in detail the influence of the nature of counter anions (Cl−, Br−, I− and NTf2−, where NTf2− = bis(trifluoromethanesulfonyl)amide) as well as the chain length of the alkyl group of the cation (1-alkyl-3-methylimidazolium or 1,3-dialkylimidazolium) in a set of ionic liquids.67 While increasing the chain length resulted in better solubility of the precursor (Et2Sb)2Te, which enhanced the formation of less aggregated nanoparticles, the strongly basic anion such as Cl− with higher coordinating capability resulted in the formation of thin Sb2Te3 nanoplates (Fig. 3e). As a consequence, the thermoelectric properties of the resulting Sb2Te3 nanoplates strongly differed. When the above precursor was decomposed in a mixture of ionic liquids containing varying amounts of Bi-containing ILs [C4mim]3[Bi3I12] and [C4mim]I, highly stoichiometric and phase-pure ternary solid solutions of the type (BixSb1−x)2Te3 could be synthesized at 150 °C (Fig. 3f).68 The synthetic details of these examples are summarized in Table 2.
| Precursor | Materials | Synthesis (temp., solvent, capping ligand, etc.) | Ref. | Comments |
|---|---|---|---|---|
| Abbreviations: C4mim = 1-butyl-3-methylimidazolium; C4C1Im = 1-butyl-3-methylimidazolium; DIPB = 1,3-diisopropylbenzene; NTf2− = bis(trifluoromethanesulfonyl)amide; ILs: ionic liquids; PVP* = poly(1-vinylpyrrolidone)graft-1-hexadecene. | ||||
| (Et2Sb)2S | Sb2S3 nanobundles | 170 °C, DIPB, PVP* | 63 | Slightly Sb-rich Sb2S3 due to formation of the volatile by-product Et2S |
| (Et2Sb)2Se | Sb2Se3 nanowires | 170 °C, DIPB, PVP* | 63 | Slightly Sb-rich Sb2Se3 due to formation of the volatile byproduct Et2Se |
| (Et2Sb)2S + (Et2Sb)2Se | Sb2(S1−xSex)3 nanowires | 170 °C, DIPB, PVP* | 63 | Impurity-free ternary phase due to excellent compatibility in the decomposition patterns of (Et2Sb)2S and (Et2Sb)2Se |
| (Et2Sb)2Te | Sb2Te3 hexagonal plates | 170 °C, DIPB, PVP* | 64 | Phase-pure product due to stoichiometric decomposition in Sb2Te3 and Et3Sb |
| (Et2Bi)2Te | Pure Bi4Te3 (80 °C), Bi4Te3 + metallic Bi (80–170 °C) | 80-170 °C, DIPB, PVP* | 65 | Metallic Bi impurity at temp. higher than 80 °C due to a weak Te–Bi bond |
| (Et2Sb)2Te | Sb2Te3 NPs | 170 °C (MW-decompos.), [C4mim]X (X = Cl−, Br−, I−, NTf2−) | 66,67 | Less agglomerated NPs by increasing the cation chain length of ILs (due to the enhanced solubility of the precursor) and thinner Sb2Te3 nanoplates by using strongly basic anions, e.g., Cl− |
| (Et2Sb)2Te + [C4mim]3[Bi3I12] | (BixSb1−x)2Te3 NPs | 150 °C (MW decompos.), [C4mim]I | 68 | Highly stoichiometric, phase-pure ternary NPs by using a Bi-containing IL |
| (Et2Bi)Te(Et) | Bi2Te NPs | 100 °C, DIPB, OA | 65 | Bi-Rich product due to a weak Te–Bi bond |
| (Ph2Sb)Te(Et) | Sb2Te3 nanoplates | 300 °C, oleylamine | 69 | High quality nanoplates at 300 °C per 2 h. A lower temp. and shorter time (250 °C per 1 h) lead to Sb2Te3 + Sb2Te mixed phases |
| (Ph2Sb)Te(Ph) | Sb2Te3 nanoplates | 250 °C, oleylamine | 69 | Perfect hexagonal nanoplates under mild conditions (250 °C per 1 h). A higher temp. and time (300 °C per 2 h) distort the shape |
| (Et2Bi)Te(Et) | Bi2Te NPs | 100 °C, DIPB or OA | 65 | Te-Poor Bi2Te particles due to formation of the volatile byproduct Et2Te |
Another related class (R2M)ER′ (M = Sb, Bi) has also been employed as SSPs for M2E3 materials. Kim et al. investigated the thermal decomposition of Ph2SbTeR (R = Et, Ph) in oleylamine.69 In the case of Ph2SbTeEt, high-quality Sb2Te3 nanoplates were obtained after heating at 300 °C for 2 h, whereas lower reaction temperatures (250 °C) and shorter reaction times (1 h) resulted in the formation of a mixture of Sb2Te as the major component and Sb2Te3 powders, indicating a rather complex decomposition mechanism. On the contrary, pure Sb2Te3 nanoplates of hexagonal shape were obtained using Ph2SbTePh at 250 °C for 1 h, whereas an increasing reaction time (2 h) and temperature (300 °C) yielded shape-distorted Sb2Te3 nanoplates. In contrast, the thermolysis of Et2BiTeEt in DIPB or OA gave pseudo-cubic Bi2Te particles, possibly due to the formation of the volatile byproduct Et2Te, leading to Te-poor Bi2Te particles.65 Interestingly, the use of a similar precursor in MOCVD led to the formation of phase-pure Bi2Te3 films on Si(100) at a very low substrate temperature of 230 °C, indicating that the decomposition mechanism for this precursor is very different in the solution and gas phases.70 EDX and XRD studies revealed the formation of highly stoichiometric and phase pure Bi2Te3 films at 215 and 230 °C, while higher substrate temperatures (245 °C) yielded Bi-rich materials with Bi4Te3 as the additional phase.
3. Bis(trialkylsilyl) monochalcogenides (R3Si)2E as facile chalcogenide-transfer reagents
The chalcogen-transfer ability of the silylated diorganyl monochalcogenides (R3Si)2E is well-known, which has allowed an easy access to metal chalcogenide nanoparticles or clusters, depending on the reaction conditions. These silylated chalcogenides (R3Si)2E react readily with a metal reagent MXn (X = alkyl, halide, amide, carboxylate, oxide), where facile and homogeneous delivery of the chalcogenide (E2−) to the metal center is promoted by the affinity of silicon(IV) for oxygen and halides as well as the volatility of the formed byproduct R3SiX (Scheme 2).71–76 When the reaction is performed in the presence of an ancillary ligand L (e.g., PR3), it often leads to the formation of high nuclear nanoclusters [MxEy(L)z].28–31 The reaction is supposed to proceed through the formation of chalcogenolate intermediates, although the high thermal instability of the M–ESiR3 bonds makes it difficult to isolate these intermediates before they afford E2−. To some extent, the stability of these chalcogenolate intermediates can be increased by using a strongly bonded ancillary ligand such as N-heterocyclic carbine (NHC) or cyclic diamine, as indicated by the isolation and structural characterization of [(IPr)M(ESiMe3)], [(tmeda)Zn(ESiMe3)2] (M = Cu, Ag; E = S, Se, Te; IPr = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene; tmeda = tetramethylethylenediamine), [(NHC)Ag–ESiMe3] (E = S, Se) and [(NHC)Ag–S–Hg–S–Ag(NHC)] at low temperature.77–79 Besides stabilizing the molecular intermediates, these ancillary ligands can also stabilize the monodisperse NPs formed at the end of the reaction, as exemplified by the synthesis of phase-pure and highly monodisperse coinage metal chalcogenide QDs (Ag2E, Cu2−xE; E = S, Se) from the room temperature reaction of (Me3Si)2E with metal halide synthons containing N-heterocyclic carbene (NHC) ligands.80 Alternatively, reagents with mixed silylated and non-silylated alkyl groups (R3Si)ER′ can be exploited to introduce surface chalcogenolate groups (Scheme 2). These ligand-protected clusters can be considered as borderline species between molecules and nanoparticles, which may serve as precursors for well-defined metal chalcogenide NPs.29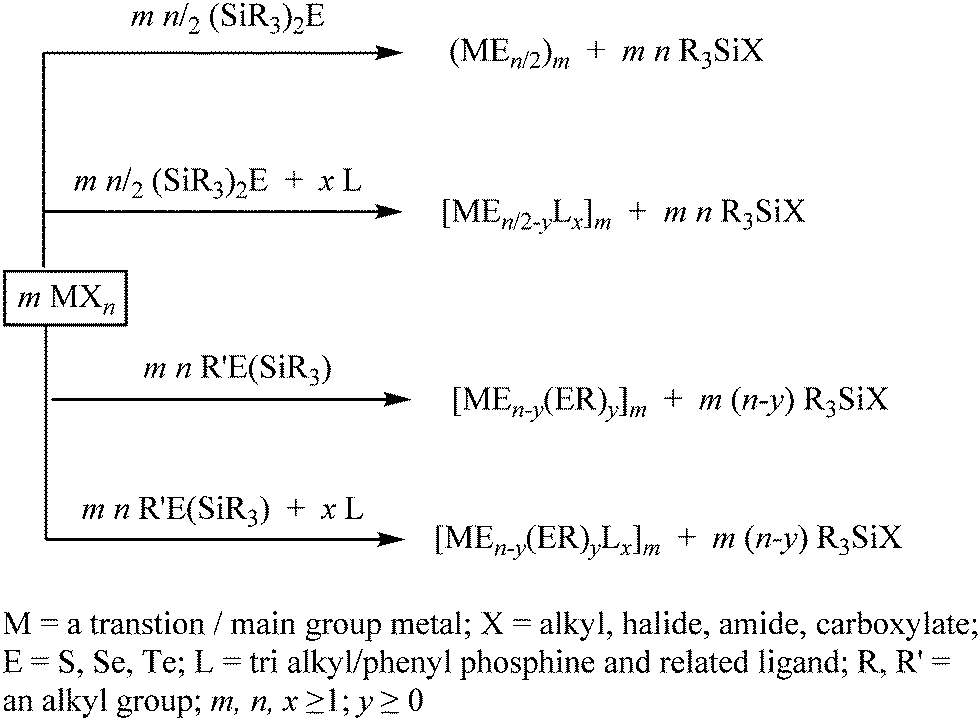 | ||
| Scheme 2 Bis(trialkylsilyl) monochalcogenides as facile chalcogenide-transfer reagents. | ||
The high reactivity of these reagents ensures not only a high yield of the products, but also a fast growth and, therefore, well-defined shape of the NPs. While the high reactivity ensures that these silylated reagents react quantitatively with the metal reagents, the good leaving property of the trialkylsilyl group makes sure that it does not act as a surface ligand (unlike other reagents such as trioctylphosphine (TOP) that can form a complex with the metal center and, therefore, limit the yield of metal chalcogenides). These properties make the bis silylchalcogenides (R3Si)2E the reagents of choice for the synthesis of metal chalcogenide NPs, despite the perceived difficulty in their handling due to high volatility and air/moisture sensitivity.31 The following subsections describe the versatility of these reagents in the synthesis of metal chalcogenide NPs under mild conditions.
3.1. Metal–organic decomposition at low-to-moderate temperatures
 | ||
| Fig. 4 Synthesis of different forms of lead chalcogenides using (Me3Si)2E as chalcogen reagents. Reproduced with permission from ref. 85–89 and 96. Copyright (2014, 2016, 2018) American Chemical Society and (2010) The Royal Society of Chemistry. | ||
When the quantity of (Me3Si)2S is less than that of the lead precursor, usually lead oleate produced in situ by the reaction of lead oxide and oleic acid (OA), the produced PbS QDs are Pb-rich with the excess of lead precursor located on the surface of the QDs and passivated by the oleic acid, leading to a stable colloidal solution.81,82 In contrast, noncolloidal aggregated suspensions of PbS NPs are formed on using an excess of (Me3Si)2S, which removes the oleic acid ligands from the surface of the particles and sulfurizes the surface.85 These S2− anions on the surface of QDs bind with the Pb atoms of adjacent QDs, thus decreasing the inter-QD spacing drastically inside the spin-coated films, leading to complete disappearance of the exciton absorption peak (Fig. 4b).
On using (Me3Si)2Se, the selenization of the QDs was also possible (i.e. the presence of Se2− instead of S2− on the surface) as confirmed by the red-shift of the absorption onset in the selenized QDs with respect to the sulfurized dots. This is in accordance with the narrower bandgap of PbSe as compared to PbS. It was noted that a chalcogen-rich termination of the PbS QD surface promoted oxidation and facilitated fusion/necking of neighboring QDs. On using PbX2 (X = Cl, Br, I) as the Pb precursors, it was possible to produce halide-terminated PbS and PbSe QDs which were air-stable (Fig. 4c).86,87 This low-temperature synthesis also helps in scaling-up of the products, as demonstrated by Zhang et al. in the synthesis of about 47 g of PbS QDs with a clear excitonic absorption peak and good size distribution within a few minutes (Fig. 4d).86
By exploiting the relative reaction rates of the chalcogenide precursors (Me3Si)2E (E = S, Se, Te), which allow for the homogeneous incorporation of the chalcogenide anions, synthesis and characterization of composition-tunable ternary lead chalcogenide alloys PbSexTe1−x, PbSxTe1−x, and PbSxSe1−x were reported (Fig. 4e).92,93 The resulting QDs were Pb-rich but the Pb/anion ratio was size- and composition-dependent and consistent with the reaction rates of the anion precursors.
Without using thiols as stabilizing ligands, Shen et al. recently synthesized HgTe colloidal quantum dots with much improved dispersion which can be stored in the cleaned solution for months without any precipitate (Fig. 5a).94 The authors suggested that the reason behind the colloidal stability was the use of a 2-fold excess of the mercury precursor. As the reaction with highly reactive (Me3Si)2Te is quantitative, there is always an excess of Hg precursor which acts as a ligand that binds to the surface Te sites. The high reactivity of (Me3Si)2Te ensures a fast growth of the particles and results in spherical quantum dots (QDs), which is in contrast with the strongly faceted HgTe QDs synthesized using TOPTe (where slow growth ensures that slowest growing facets dominate the final structures).
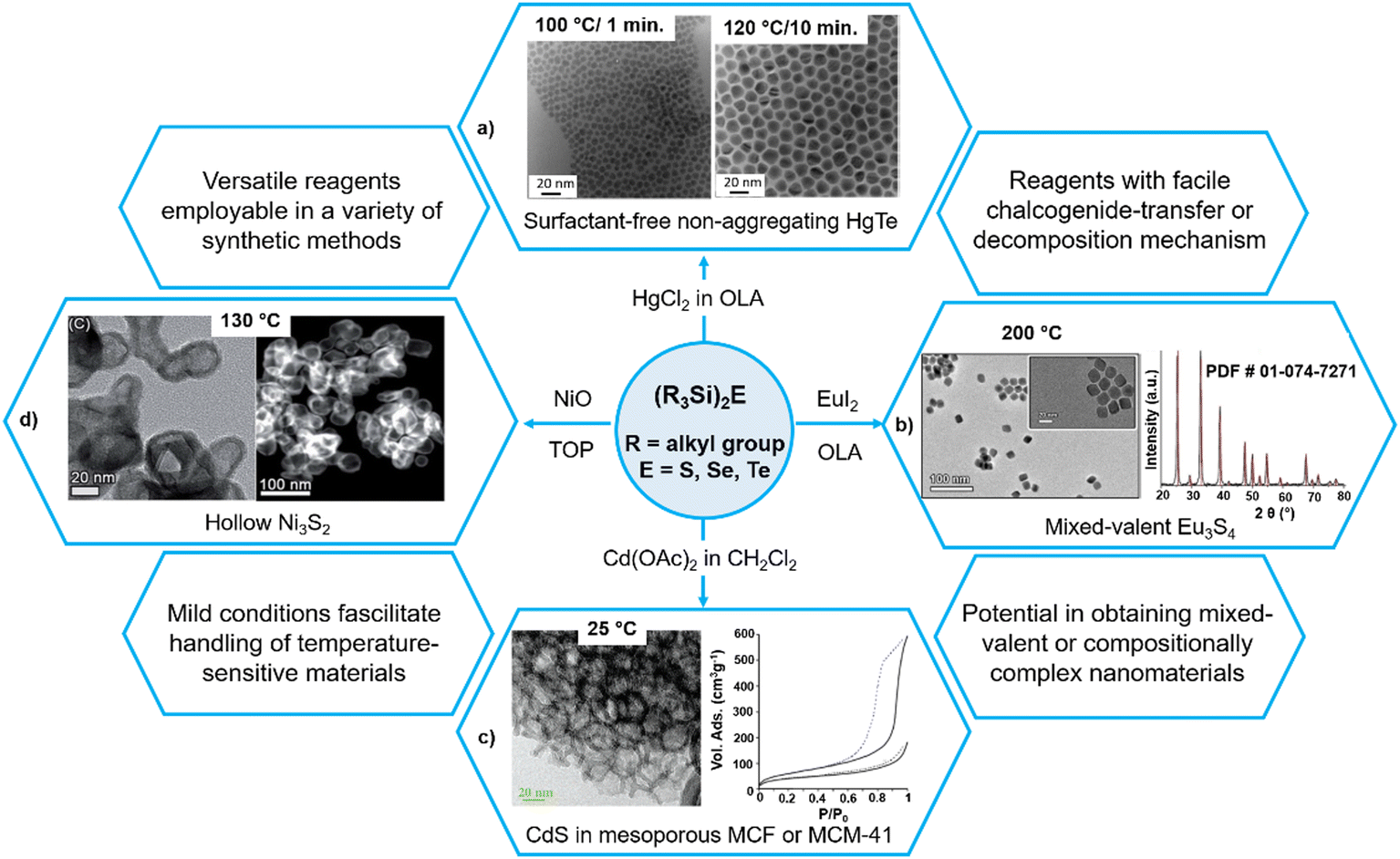 | ||
| Fig. 5 Versatility of (Me3Si)2E in the mild synthesis of different nanoforms of metal chalcogenides. Reproduced with permission from ref. 94, 95, 107 and 113. Copyright (2007, 2017) American Chemical Society; (2017) The Royal Society of Chemistry; and (2021) Wiley-VCH. | ||
By reacting (Me3Si)2S with divalent lanthanide halides LnX2 (X = Cl, Br, I) in oleylamine, Stoll et al. recently synthesized mixed-valent lanthanide sulfide Eu3S4 and EuSm2S4 NPs at 200 °C (Fig. 5b).95 The authors reported the combination of the high reactivity of (Me3Si)2S and the use of low-valent metal halides as a key to stabilize the mixed-valent phases. The fact that only EuI2 afforded phase-pure Eu3S4 (the use of EuCl2 gave EuS NPs, whereas EuBr2 yielded a contaminated sample) highlights the influence of the choice of reagent and the role of kinetics in phase stabilization.
The reaction of (R3Si)2Te (R = Me, Et) with GeX2 (X = chloride, aminoalkoxide) afforded GeTe NPs at low temperatures.97 Specifically, the reaction of (Et3Si)2Te and GeCl2·dioxane was studied in detail. The presence of protic and strongly coordinating primary or secondary amine solvents was essential for the formation of GeTe NPs with defined chemical composition, size, and shape. The nuclear magnetic resonance (NMR) studies revealed that the reaction in oleylamine (OLA) did not proceed with the elimination of Et3SiCl. Rather, OLA reacted with (Et3Si)2Te at ambient temperature with the formation of first silylamine (oleylN(H)SiEt3) and then the [Te4]2− dianion, which then reacted with GeCl2·dioxane to afford GeTe at temperatures below 150 °C. In contrast, reactions in 1,3-diisopropylbenzene occurred with the elimination of Et3SiCl and subsequent formation of agglomerated GeTe particles, whereas reactions in tri-n-octylamine and 1-hexadecanethiol produced elemental tellurium. In the presence of an excess of (Et3Si)2Te, GeTe NPs decorated with Te nanowires were obtained. Similarly, the reactions of (R3Si)2E (R = Me, Et; E = S, Se, Te) with metal aminoalkoxides of Sb(III) and Bi(III) at moderate temperatures afforded phase-pure M2E3 NPs without any additional elemental or metal-rich phases.97,98
Ternary nanoparticles containing two different metals, i.e., AgSbS2 NPs, were synthesized by reacting simultaneously AgNO3 and SbCl3 or [Sb(diethyldithiocarbamate)3] with (Me3Si)2S in a mixture of OLA and OA at 180–220 °C.99 Upon varying the reaction time between 1.5 and 5 minutes, the reaction temperature between 180 and 220 °C or the OLA/OA volume ratio in the range of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 to 7.25:0.25, a wide range of various sized AgSbS2 NPs with narrow size distributions were obtained in a very precise manner. Similarly, ternary AgInSe2 NPs were synthesized by the hot-injection method by injecting (Me3Si)2Se into a solution of AgI and InI3 in a mixture of OA and TOP at 280 °C.100 The reaction of (Me3Si)2S with CuIn(OH)5 in a mixture of 1-octadecene (ODE), OA and OLA yielded OA-capped CuInS2 NPs at 200 °C, where the use of a bimetallic precursor containing a 1:1 ratio of the two metals ensured the desired composition in the synthesized ternary NPs.101
:4 to 7.25:0.25, a wide range of various sized AgSbS2 NPs with narrow size distributions were obtained in a very precise manner. Similarly, ternary AgInSe2 NPs were synthesized by the hot-injection method by injecting (Me3Si)2Se into a solution of AgI and InI3 in a mixture of OA and TOP at 280 °C.100 The reaction of (Me3Si)2S with CuIn(OH)5 in a mixture of 1-octadecene (ODE), OA and OLA yielded OA-capped CuInS2 NPs at 200 °C, where the use of a bimetallic precursor containing a 1:1 ratio of the two metals ensured the desired composition in the synthesized ternary NPs.101
3.2. Use of ionic liquids
Ionic liquids offer an interesting synthetic route, not only as effective solvent media, but also as stabilizers and shape-directing templates. Thus, (Et3Si)2Se reacted with SbCl3 in [C4C1Im]Cl (C4C1Im = 1-butyl-3-methylimidazolium) and oleic acid to give phase-pure Sb2Se3 at 150 °C as agglomerated nanowires with a high aspect ratio.102 Similarly, reactions of (Et3Si)2E with bismuth-containing ionic liquids [Cat]3[Bi3I12] [Cat = C4C1Im or C4mim] in the presence of oleylamine at 150 °C afforded phase-pure Bi2E3 (E = Se, Te).68,102,103 Alternatively, these silylated organyl chalcogenides (R3Si)2E can easily be converted to chalcogenide-containing ionic liquids Cat+[ESiMe3]− (Cat+ = an organic cation) of comparable activity but with higher thermal stability, therefore effectively suppressing the undesired homolytic cleavage of the E–Si bond observed occasionally in the parent (R3Si)2E.104 For instance, [C4C1Pyr][ESiMe3] (C4C1Pyr = 1-butyl-1-methylpyrrolidinium; E = Se or Te) reacts with bismuth-containing [C4C1Im]3[Bi3I12] in [C4C1Im]I at 150 °C to afford phase-pure Bi2E3 free from any elemental Se/Te impurities.1053.3. Sophisticated and intricate multicomponent architectures
One important advantage of the highly reactive nature of these silylated chalcogenides is that they generate metal chalcogenide NPs under mild conditions and therefore can be exploited to generate composites with sensitive materials. For example, these have been employed on a modified mesoporous surface such as Mobil Composition of Matter no. 41 (MCM-41) and mesocellular silica foam (MCF) to form binary CdE and ZnE or ternary CdxZn1−xE (E = S, Se, Te) nanomaterials within the host framework at room temperature, and therefore without modifying its mesoporous characteristics (Fig. 5c).106,107 Even though the surface area of the host matrix after loading with NPs diminishes, it still remains sufficiently high (e.g., 250 m2 g−1 in CdE loaded MCF against 715 m2 g−1 in MCF). More importantly, the resulting materials maintain their mesoporosity, indicating their suitability for many applications.These synthons have also been employed to get sophisticated core@shell structures. For example, the reaction of the pre-formed Cu2−xSe NPs with ZnEt2 and (Me3Si)2S in a mixture of OLA and trioctylphosphine oxide (TOPO) afforded core@shell Cu2−xSe@ZnS NPs at 175 °C.108 Similarly, the reaction of ZnO tripods with (Me3Si)2S in TOP/TOPO at 250 °C yielded hollow ZnS tripods which on partial cation exchange with Cd2+ at 220 °C afforded ZnS@CdS core–shell tripod heterostructures.109
3.4. Miscellaneous methods
As said earlier, these ligands can react with non-conventional reagents under a variety of conditions. For instance, using the microemulsion method, nanocrystalline Bi2Te3 with particle sizes less than 10 nm were synthesized from the reaction of BiO(ClO4) with (Me3Si)2Te in a reverse micelle formed by sodium dioctyl sulfosuccinate and water in hexane in 70% yield.110 Similarly, these ligands can also react with pre-formed metallic or metal oxide nanoparticles. For example, (Me3Si)2Te reacts with pre-formed colloidal metal nanoparticles of Pd, Pt and Ni under sufficiently mild conditions (180 °C, 1 h) to form their corresponding ditellurides PdTe2, PtTe2, and NiTe2.111 Other metal nanoparticles of Co, Ag, and Rh also form crystalline metal tellurides upon reaction with (Me3Si)2Te, indicating that this approach to synthesizing nanoscale transition metal tellurides is general. Interestingly, the reactions of (Me3Si)2E with pre-formed nanocrystalline metal oxides (CdO, ZnO, NiO, Co3O4 and CoO) at 135–235 °C in TOP/TOPO resulted in nanocrystalline metal chalcogenides (CdS, ZnS, Ni3S2, Ni5Se5, Co9S8 and Co3Se4) with hollow, vesicle-like morphologies, as confirmed by transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images (Fig. 5d).108,112,113 The shape, single crystallinity, and orientation of the parent metal oxide NPs are completely preserved in the transformed particles. The authors explained the hollow morphologies on the basis of a nanoscale Kirkendall-type effect, arising from differential ion diffusion rates between ions in the reactant and product phases. All nanocrystalline chalcogenides produced were surrounded by an outer amorphous silica layer, which could help in preventing agglomeration and degradation. These organyl chalcogenides can also react with more complex precursors such as mixed-metal oxides to yield ternary metal chalcogenide NPs as demonstrated by the reaction of NiCo2O4 with (Me3Si)2S which generated NPs of heterobimetallic (Ni/Co)9S8 without significant phase segregation.113Table 3 summarizes some selected examples of metal chalcogenide nanomaterials synthesized under mild conditions using silylated organyl chalcogenides.| Reagents/precursors | Materials | Synthesis (temp., solvent, capping ligand, etc.) | Ref. | Comments |
|---|---|---|---|---|
| Abbreviations: AOT = sodium dioctyl sulfosuccinate; C4C1Im = 1-butyl-3-methylimidazolium; DIPB = 1,3-diisopropylbenzene; dmea = 1,1-dimethylethanolamine; DMF = dimethylformamide; OA = oleic acid; ODE = 1-octadecene; ODT = octadecane thiol; OLA = oleylamine; TOP = trioctylphosphine; TOPO = trioctylphosphine oxide; MW = microwave; RT = room temperature; NHC = N-heterocyclic carbene; NPs = nanoparticles; QDs = quantum dots. | ||||
| Binary metal chalcogenides | ||||
| Pb(oleate)2 in excess + (Me3Si)2S | Pb-Rich PbS QDs | 115 °C, OA, ODE | 81 and 82 | Stable colloidal solution due to the presence of oleic acid-protected lead ions on the surface of QDs |
| PbI2 + (Me3Si)2E in excess | E-Stabilized PbS NPs (E = S, Se) | 125 °C, DMF, OA, ODE, OLA | 85 | The E2− anions on the surface of QDs bind with the Pb atoms of adjacent QDs, which decrease the inter-QD spacing and lead to complete disappearance of the exciton absorption peak |
| PbX2 (X = Cl, Br, I) + (Me3Si)2E | Halide-stabilized and air-stable PbE (E = S, Se) QDs | 100–140 °C, OLA | 86 and 87 | Facile scaled-up synthesis (∼47 g of high-quality PbS QDs within a few min) |
| Pb(oleate)2 + (Me3Si)2S | Olive oil-stabilized PbS NPs | 60 °C, olive oil, OA, ODE | 88 | Use of olive oil as a green solvent lowers further the synthetic temp. and makes NPs more air-stable |
| Pb(oleate)2 + (Me3Si)2S | PbS nanosheets | 90 °C, Ph2O, TOP, 1,1,2-TCE, DMF | 89 | Use of 1,1,2-TCE and DMF essential for getting nanosheets |
| GeCl2·dioxane + (Et3Si)2Te | GeTe NPs | 160 °C, OLA | 97 | Oleylamine essential for the formation of GeTe NPs with defined chemical composition, size, and shape |
| M(dmea)3 (M = Sb, Bi) + (R3Si)2E (R = Me, Et; E = S, Se, Te) | M2E3 NPs (M = Sb, Bi) | 120-140 °C, DIPB | 98 | Thermally stable but chemically reactive metal aminoalkoxides lead to phase-pure Bi2Te3 without any Bi impurity |
| BiO(ClO4) + (Me3Si)2Te | Bi2Te3 NPs | RT, reverse micelles in AOT, H2O and hexane | 110 | The microemulsion method facilitates RT synthesis of monodisperse, sub-10 nm NPs |
| SbCl3 + (Et3Si)2Se | Sb2Se3 nanowires | 150 °C, OLA, [C4C1Im]Cl | 102 | Reaction easily scalable up to 10 g of NPs |
| (Et3Si)2E + [C4C1Im]3[Bi3I12] or [C4mim]3[Bi3I12] | Bi2E3 (E = Se, Te) | 150 °C, OLA, [C4C1Im]Cl or [C4mim]I | 68, 102 and 103 | Oleylamine effectively suppresses the homolytic Se–Si bond cleavage to afford the phase-pure product |
| Zn(OAc)2(3,5-Me2Py)2 + (Me3Si)2E (E = S, Se, Te) + MCM-41 | ZnE NPs (E = S, Se, Te) within MCM-41 | RT, toluene, CH2Cl2 | 106 | RT synthesis ensures no modification of the mesoporous characteristics of MCM-41 |
| Cd(OAc)2(PnBu3)2 + (Me3Si)2E (E = S, Se, Te) + MCM-41/MCF | CdE (E = S, Se, Te) within the mesoporous hosts | RT, CH2Cl2 | 107 | The organic ligand, which anchors the CdE material within MCM-41/MCF, is cleaved on heating beyond 135 °C, resulting in the aggregation of CdE outside of the pores |
| HgCl2 in excess + (Me3Si)2Te | Non-aggregating HgTe QDs | 100–120 °C, OLA | 94 | Use of an excess of HgCl2 ensures a well-dispersed and stable colloidal suspension |
| EuCl2 + (Me3Si)2S | EuS NPs | 200 °C, OLA | 95 | — |
| EuI2 + (Me3Si)2S | Mixed-valent Eu3S4 | 200 °C, OLA | 95 | Initially Eu S is formed at lower temperatures, which is converted to Eu3S4 over time |
| SmI2 + (Me3Si)2S | Mixed-valent Sm3S4 | 300 °C, OLA | 95 | Higher reaction temperature due to the greater redox potential of samarium |
| EuI2 + SmI2 + (Me3Si)2S | Mixed-valent EuSm2S4 | 300 °C, OLA | 95 | Based on XPS, the authors described the materials as (Eu2+)(Eu3+,Sm3+)2S4 |
| (NHC)MBr (M = Cu, Ag) + (Me3Si)2S | NHC-protected Cu2−xS or Ag2S QDs | RT, CH2Cl2, ODE | 80 | Formation of NHC–Ag2S QDs via the intermediate complex [NHC–AgSAg–NHC] |
| (NHC)MBr (M = Cu, Ag) + (tBuMe2Si)2Se | NHC-protected Cu2−xSe or Ag2Se QDs | RT, CH2Cl2, THF | 80 | NHC ligands containing long carbon chains have superior competence over commonly used organic capping ligands (OA, OLA) for obtaining highly monodisperse QDs |
| Metal NPs (Pd, Pt, Ni) + (Me3Si)2Te | MTe2 NPs (M = Pd, Pt, Ni) | 180 °C, TOPO, OLA | 111 | The high reactivity of silylated (Me3Si)2Te ensures formation of the telluride-rich phase MTe2 over MTe |
| (Me3Si)2S + ZnO NPs | ZnS NPs with hollow morphology | 235 °C, TOP/TOPO | 112 | The tetrapod morphology of ZnO is preserved in ZnS |
| (Me3Si)2S + CdO NPs | CdS NPs with hollow morphology | 130 °C, TOP/TOPO | 109 | The pseudo-spherical shape and crystallinity of CdO are preserved in the CdS NPs |
| (Me3Si)2E (E = S, Se) + NiO NPs | Ni3S2 or Ni5Se5 NPs with hollow morphology | 130 °C, TOP | 113 | Silylated organyl chalcogenides generate a protecting amorphous silica layer around NPs |
| Co3O4 or CoO NPs + (Me3Si)2E (E = S, Se) | Co9S8 or Co3Se4 NPs with hollow morphology | 130 °C, TOP | 113 | Hollow morphologies due to the nanoscale Kirkendall-type effect arising from different ion diffusion rates |
| Cu(TFA)2 + (Me3Si)2S | ODT-capped CuS NPs | RT, ODT, Et2O | 43 | The high reactivity of (Me3Si)2S ensures the formation of phase-pure CuS in 90% yield |
| Ag(TFA) + (Me3Si)2S | ODT-capped Ag2S NPs | RT, ODT, Et2O | 43 | Instant formation of phase-pure Ag2S in 95% yield |
| Ternary metal chalcogenides | ||||
| Pb(oleate)2 + (Me3Si)2E (E = S, Se, Te) | PbSexTe1−x, PbSxTe1−x, and PbSxSe1−x | 140 °C, OA and ODA | 92 | Composition-tunable ternary alloys due to excellent compatibility among (Me3Si)2E |
| Pb(oleate)2 + (Me3Si)2S + (Me3Si)2Se | PbSxSe1−x (x = 0.2–0.8) | 130 °C, olive oil, ODA | 93 | Comparable reactivities of (Me3Si)2S and (Me3Si)2Se allow homogeneous incorporation of the chalcogenide anions |
| AgNO3 + SbCl3 + (Me3Si)2S | AgSbS2 NPs | 180–220 °C, OLA, OA | 99 | Various sized AgSbS2 NPs with narrow size distributions as a function of reaction conditions (time, temp. or the OLA/OA volume) |
| (Me3Si)2S + NiCo2O4 NPs | (Ni/Co)9S8 NPs with hollow morphologies | 130 °C, TOP | 113 | High reactivity of silylated (Me3Si)2S ensures no significant phase segregation |
| CuIn(OH)5 +(Me3Si)2S | OA-Capped CuInS2 NPs | 200 °C, ODE, OA, OLA | 101 | NPs remain well-dispersed in non-polar solvents for several months |
| Cu2−xSe NPs + ZnEt2 + (Me3Si)2S | Core@shell Cu2−xSe@ZnS NPs | 175 °C, OLA, TOPO | 108 | Stable plasmonic behavior by the core–shell structure |
4. Diorganyl dichalcogenides
Since their first use in the synthesis of SnSe and SnTe nanoparticles in 2002,114 diorganyl dichalcogenides R2E2 have emerged as facile chalcogenide reagents in the synthesis of binary, ternary and multinary metal chalcogenide NPs of controlled size and shape.26,274.1. A comparison with diorganyl monochalcogenides
These reagents enjoy mostly the same advantages described above for the R2E reagents, i.e. easy tunability of reactivity by varying the group R, facile transfer of the chalcogenide anion under mild conditions, easily removable byproducts, good solubility in common organic solvents, etc. However, these diorganyl dichalcogenides R2E2 do have an additional E–E bond which is cleaved rather easily under mild conditions. As a result, they rarely act as neutral ligands towards metal centres and, unlike a rich library available of metal complexes with organyl monochalcogenides,32,33 there are very few metal complexes reported so far with neutral R2E2 ligands, mostly isolated at a low temperature.115,116 However, these complexes are not stable and quickly decompose to give monochalcogenide complexes.115,116 While the presence of an additional E–E bond alters the decomposition pathways of R2E2 and makes them slightly less reactive than R2E reagents, it also renders them more air- and temperature-stable, resulting in easy handling and more commercial availability of a wide variety of differently substituted diorganyl dichalcogenides R2E2 (where E = S, Se, or Te and R = alkyl, allyl, benzyl, or aryl).25–27 Another advantage of these reagents is that they can easily be transformed in situ to monoalkyl chalcogenides R–E via E–E cleavage, which can act as capping ligands to stabilize the formed NPs. This has implications in the chemistry of RSe- and RTe-stabilized NPs as, unlike thiol ligands, which have been extensively employed as surfactant for semiconducting NPs, the selenol and tellurol ligands are usually oxidatively instable and hence are difficult to use.26 With an exception of a couple of reports,117,118 the diorganyl dichalcogenides employed so far in the synthesis of metal chalcogenides are almost entirely non-silylated. This is in contrast to diorganyl monochalcogenides where silylated derivatives form the bulk of the reported examples.The decomposition of R2E2, either thermally25–27 or photolytically,119,120 proceeds through the radical scission of the E–E and E–C bonds. The mechanism has been studied in detail by Vela et al. who carried out a combined experimental and DFT study correlating the bond strengths in R2E2 with the size and shape of the obtained CdSe and CdS NPs.25 They demonstrated that, by changing the R group within the series of R2E2 (E = S, Se), the reactivities of these chalcogenide reagents could be tuned. While the E–E bond strength was found to be relatively constant, the calculated C–E bond strengths changed significantly upon substituting the R group and determined the overall reactivity of the R2E2. As compared to the R2E2 with weaker C–E bonds which were reactive and afforded isotropic NPs, those with large C–E bond dissociation energies were less reactive and yielded anisotropic, and often morphologically metastable NPs with higher surface areas (Fig. 6). Interestingly, the strength of the C–Se bond not only influences the kinetics of R2E2, but also determines the nature of the crystalline phase and composition of the product. For instance, the reactions of diallyl disulfide [(allyl)2S2] and dibenzyl disulfide with FeCl2 in oleylamine at 220 °C yielded the pyrite (FeS2) and greigite (Fe3S4) phases, respectively, whereas the reactions of di-tert-butyl disulfide tBu2S2 and di-phenyl disulfide (Ph2S2) under similar conditions produced the pyrrhotite (Fe7S8) phase.27 By exploiting this easy tunability of the reactivities by varying the R group, a number of articles have described the use of differently substituted R2E2 in the synthesis of binary, ternary or even multinary metal chalcogenide NPs in the last ten years. Some selected examples are summarized in Table 4.
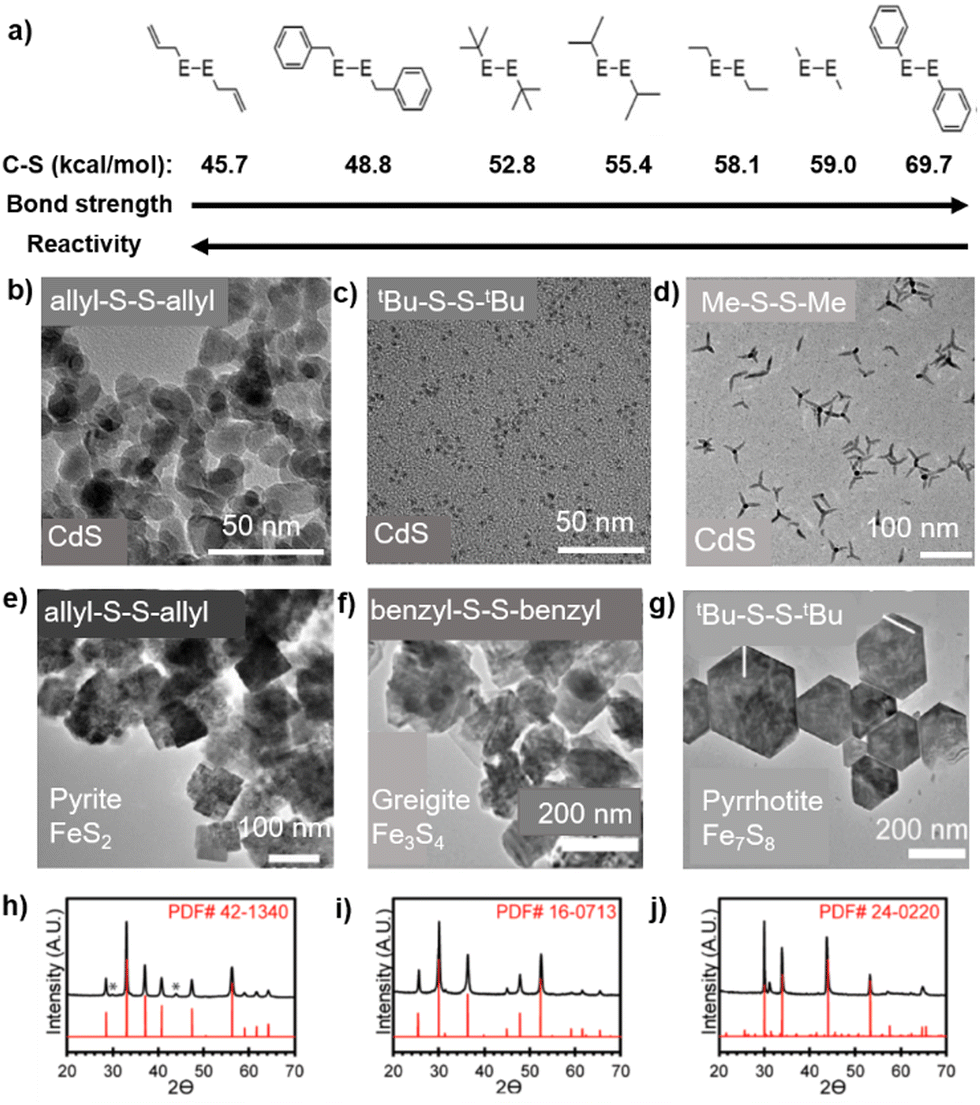 | ||
| Fig. 6 Varying C–S bond strengths and reactivities among R2E2 precursors (a) and its influence on the particle shape (b–d) as well as composition and crystalline phase (e–j) of metal chalcogenide nanomaterials. Adapted with permission from ref. 25 and 27. Copyright (2013, 2017) American Chemical Society. | ||
| Reagents | Materials | Synthesis (temp., solvent, capping ligand, etc.) | Ref. | Comments |
|---|---|---|---|---|
| Abbreviations: DDA = dodecylamine; HAD = hexadecylamine; DDT = dodecanethiol; HMDS = hexamethyldisilazane (Me3Si)2NH; OA = oleic acid; ODE = 1-octadecene; ODT = octadecane thiol; OLA = oleylamine; TOP = trioctylphosphine; TOPO = trioctylphosphine oxide; RT = room temperature; NPs = nanoparticles. | ||||
| Ph3Bi + tBu2Se2 | BiSe NPs | RT photolysis (254 nm per 4 h), DDA, P-123 copolymer | 120 | Ph3Bi and tBu2Se2 both undergo homolytic photolysis without using a reducing reagent to produce 5 nm BiSe NPs |
| GeI4 + Ph2Se2 | GeSe nanosheets | 268 °C, HDA | 121 | — |
| Metallic Sn NPs + Ph2Te2 | Surfactant-free SnTe NPs | 165 °C, diglyme | 114 | Large spherical NPs (∼60 nm) or smaller star-shaped agglomerates of needles (∼15 × 40 nm) are obtained depending on the Sn/Ph2Te2 ratio |
| SnCl2 + tBu2Se2 | SnSe NPs | 180 °C, DDA, DDT | 122 | Stoichiometric SnCl2/tBu2Se2 ratio yielded phase-pure SnSe, whereas an excess of tBu2Se2 yielded SnSe2 |
| SnI4 + GeI4 + tBu2Se2 | Ternary SnxGe1-xSe NPs | 225 °C, DDA, HMDS | 123 | In the absence of HMDS, only binary SnSe NPs were produced |
| CuCl + SnI4 + tBu2Se2 | Wurtzite Cu2SnSe3 NPs | 180 °C, DDA, DDT | 42 | The combination of DDT and tBu2Se2 is critical to obtain the wurtzite phase of Cu2SnSe3 |
| In(acac)3 + tBu2S2 | In2S3 nanorods | 180 °C, OLA | 124 | Synthesis of In2S3 nanorods takes 7 h due to the less reactivity of tBu2S2 |
| CuCl + In(acac)3 + tBu2S2 | Wurtzite Cu–In–S NPs | 180 °C, OLA, DDT | 125 | Slow formation of In2S3 leads to the isolation of a metastable wurtzite Cu–In–S phase |
| Cu(oleate)2 + Ph2Se2 | Wurtzite-like CuInSe2 NPs | 220 °C, OLA | 49 | The metastable phase is formed through the Cu3Se2 intermediate |
| InCl3 + SnCl2 + Ph2Se2 | In4SnSe4 microwires | 280 °C, HMDS, OLA, ODE | 126 | — |
| ZnO + tBu2S2 | ZnS NPs | 235 °C, OLA, TOP | 127 | ZnS with a hollow structure, induced by the nanoscale Kirkendall effect |
| Cd(oleate)2 + R2E2 (R = allyl, Bz, tBu, iPr, Et, Me, Ph; E = S or Se) | CdS and CdSe NPs | 250 °C, OLA, OA, ODE | 25 | CdS and CdSe NPs with spherical, elongated or tetrapodal morphology by varying the reactivity of R2E2 |
| CuCl2 + tBu2S2 | Cu2−xS NPs | 180-220 °C, OLA + DDT | 128 | Roxbyite Cu1.78S or Djurleite Cu1.96S depending on Cu concentration |
| Cu(acac)2 + (dodecyl)2Se2 | Wurtzite Cu2−xSe NPs | 155 °C, ODE | 50 | Metastable wurtzite Cu2−xSe NPs are irreversibly transformed to thermodynamic cubic Cu2Se at 185 °C |
| Cu(oleate)2 + Ph2Se2 | Wurtzite Cu3Se2 NPs | 220 °C, OLA | 49 | Metastable wurtzite Cu3Se2 NPs |
| Cu(acac)2 + (dodecyl)2Te2 | Pseudo-cubic Cu1.5Te NPs | 135 °C in a variety of solvents (ODE, OlA, OA and DOE) | 51 | Metastable pseudo-cubic Cu1.5Te NPs are formed at 135 °C, whereas a thermodynamic vulcanite CuTe phase is obtained at 185 °C |
| Cu(oleate)2 + Ph2Se2 + Zn(oleate)2 + Fe(acac)3 | Wurtzite-like Cu2FeSnSe4 NPs | 250 °C, OLA | 129 | Metastable quaternary NPs through sequential cationic replacement in the initially formed binary Cu3Se2 NPs |
| Cu(oleate)2 + Ph2Se2 + Zn(oleate)2 + Sn(ethylhexanoate)2 | Wurtzite-like Cu2ZnSnSe4 NPs | 255 °C, OLA | 130 | Metastable quaternary NPs through sequential cationic replacement in the initially formed binary Cu3Se2 NPs |
| AgNO3 + In(OAc)3 + Bz2Se2 | Metastable AgInSe2 NPs | 230 °C, OLA, OA, ODE | 131 | Presence of oleylamine essential to get the metastable phase |
| CuCl + Fe(acac)3 + Ph2Se2 | CuFeSe2 nanosheets | 255 °C, OA, OLA, ODE | 132 | Hierarchical nanostructures assembled from nanosheets of ∼50 nm thickness and ∼400 nm side length |
| FeCl2 + (allyl)2S2 | Pyrite FeS2 NPs | 220 °C, OLA | 27 | Due to a lower C–S bond energy as compared to the S–S one, (allyl)2S2 releases a persulfide (S–S)2− ion, leading to formation of pyrite FeS2 NPs |
| FeCl2 + (benzyl)2S2 | Greigite Fe3S4 NPs | 220 °C, OLA | 27 | A sulphur-deficient phase is obtained through the rupture of the S–S bond |
| FeCl2 + R2S2 (R = tBu, Ph) | Pyrrhotite Fe7S8 NPs | 220 °C, OLA | 27 | A sulphur-deficient phase is obtained through the rupture of the S–S bond |
| WCl4 + tBu2Se2 | Amine-stabilized WSe2 nanosheets | 225 °C, DDA | 133 | Addition of tetra-n-octylammonium bromide prevents agglomeration of colloidal 2H-WSe2 nanosheets |
| W(CO)6 + Ph2Se2 | WSe2 NPs | 330 °C, OA, TOPO | 134 | A higher OA/W(CO)6 molar ratio yields slower nucleation, larger NPs and a shift from the 2H to the 1T' crystal phase |
| W(CO)6 + Ph2Se2 | MSe2 (M = Mo, W) nanosheets | 330 or 350 °C, OA, OLA, oleyl alcohol | 135 | Single-layer MSe2 nanosheets obtained by using oleic acid, while multilayer nanosheets are formed with oleyl alcohol and oleylamine |
| MoCl5 + WCl6 + Ph2Se2 | MoxW1−xSe | 300 °C, OA, OLA, HMDS | 136 | Easily tunable compositions by varying the Mo and W ratio |
The different reactivities of diorganyl mono- and dichalcogenides can be highlighted by comparing the reactions of tBu2E and tBu2E2. As compared to tBu2E which often affords molecular precursors with low decomposition temperature (Fig. 1) or even NPs at room temperature (Fig. 2), the reaction of tBu2E2 requires relatively high temperature to afford NPs of binary and ternary metal chalcogenide NPs (Fig. 7). For instance, tBu2Se2 reacts with SnCl2 or a combination of CuCl and SnI4 to yield binary SnSe and ternary Cu2SnSe3 NPs, respectively, at 180 °C in the presence of dodecylamine (DDA) and dodecanethiol (DDT) (Fig. 7a and b).42,122 While the stoichiometric ratio of Sn and Se reagents was essential for getting phase-pure SnSe NPs, the use of DDT and hexamethyldisilazane (HMDS) was critical to obtain the new wurtzite phase of Cu2SnSe3 NPs. Similarly, ternary SnxGe1−xSe NPs were prepared by reacting tBu2Se2 with varying ratios of GeI4 and SnI4 in DDA and HMDS at 225 °C.123 The decomposition of tBu2E2 (E = Se, Te) can be triggered photolytically also, thus decreasing the reaction temperature significantly.119,120 For example, the photolytic decomposition at 254 nm for 4 h of the deaerated solution of Ph3Bi and tBu2Se2 in water containing dodecylamine and a triblock copolymer P-123 yielded BiSe NPs of an average size of 5 nm (Fig. 7c).120 As observed in the case of the R2E series, a stronger C–S bond (in comparison to the C–Se bond) makes R2S2 slightly less reactive. For example, the reaction of CuCl2 with an excess of tBu2S2 yields roxbyite Cu2−xS NPs, where a slow but continuous growth of the nanoparticles (due to the low nucleation rate of the reagent) not only ensured accurate size control of the NPs but also allowed to follow their gradual morphology evolution from spheres and disks to tetradecahedrons and dodecahedrons (Fig. 7d).128 The relatively slow reactivity of tBu2S2 is further manifested from its reaction with In(acac)3 which required 7 h to afford In2S3 at 180 °C (Fig. 7e).124 This slow rate was subsequently exploited to synthesize metastable wurtzite ternary CuInS NPs by performing the above reaction in the presence of CuCl and DDT (Fig. 7f).125 Other sulphur sources such as elemental S8 or tert-butyl thiol produced either no crystalline products or thermodynamic chalcopyrite phase.
 | ||
| Fig. 7 Utilization of tBu2E2 (E = S, Se) precursors to get binary and ternary metal chalcogenide nanoparticles. Adapted with permission from ref. 42, 120, 122, 124, 125, and 128. Copyright (2011) The Royal Society of Chemistry and (2009, 2009, 2010, 2011, 2012) American Chemical Society. | ||
4.2. Isolation of metastable phases
While materials in metastable crystal structures can afford useful properties, synthesizing metastable materials necessarily requires kinetic control of the chemistry away from thermodynamic equilibrium, which is not always easy to achieve. The above results showed that variable reactivity in the R2E2 series, obtained simply by changing the R and E, could be utilized to control the reaction kinetics and hence predict and provide the synthetic pathways for thermodynamically or kinetically driven products. Unsurprisingly, these reagents have been employed prominently in the past few years for the synthesis and isolation of several metastable phases (Fig. 8). For instance, the reactions of Cu(acac)2 with (dodecyl)2E2 (E = Se, Te) in a variety of solvents (ODE, OlAm, OA or DOE) afforded metastable wurtzite Cu2−xSe and pseudo-cubic Cu1.5Te NPs at 155 and 135 °C, respectively.50,51 The right choice of the organyl group in R2E2 was important for isolating these metastable phases under mild synthetic conditions because at higher temperature (185 °C) these phases are irreversibly transformed into the thermodynamic cubic Cu2Se and vulcanite CuTe phases, respectively. Similarly, using Ph2Se2 that possesses a higher C-Se bond strength, a “wurtzite-like” metastable phase of the ternary CuInSe2 was obtained, whereas R2Se2 (R = methyl, benzyl) with weaker C–Se bonds yielded NPs with the thermodynamic chalcopyrite crystal structure.49 These two phases are formed via distinct copper selenide intermediates, with the wurtzite-like phase forming through Cu3Se2 and the chalcopyrite forming through Cu2−xSe intermediates.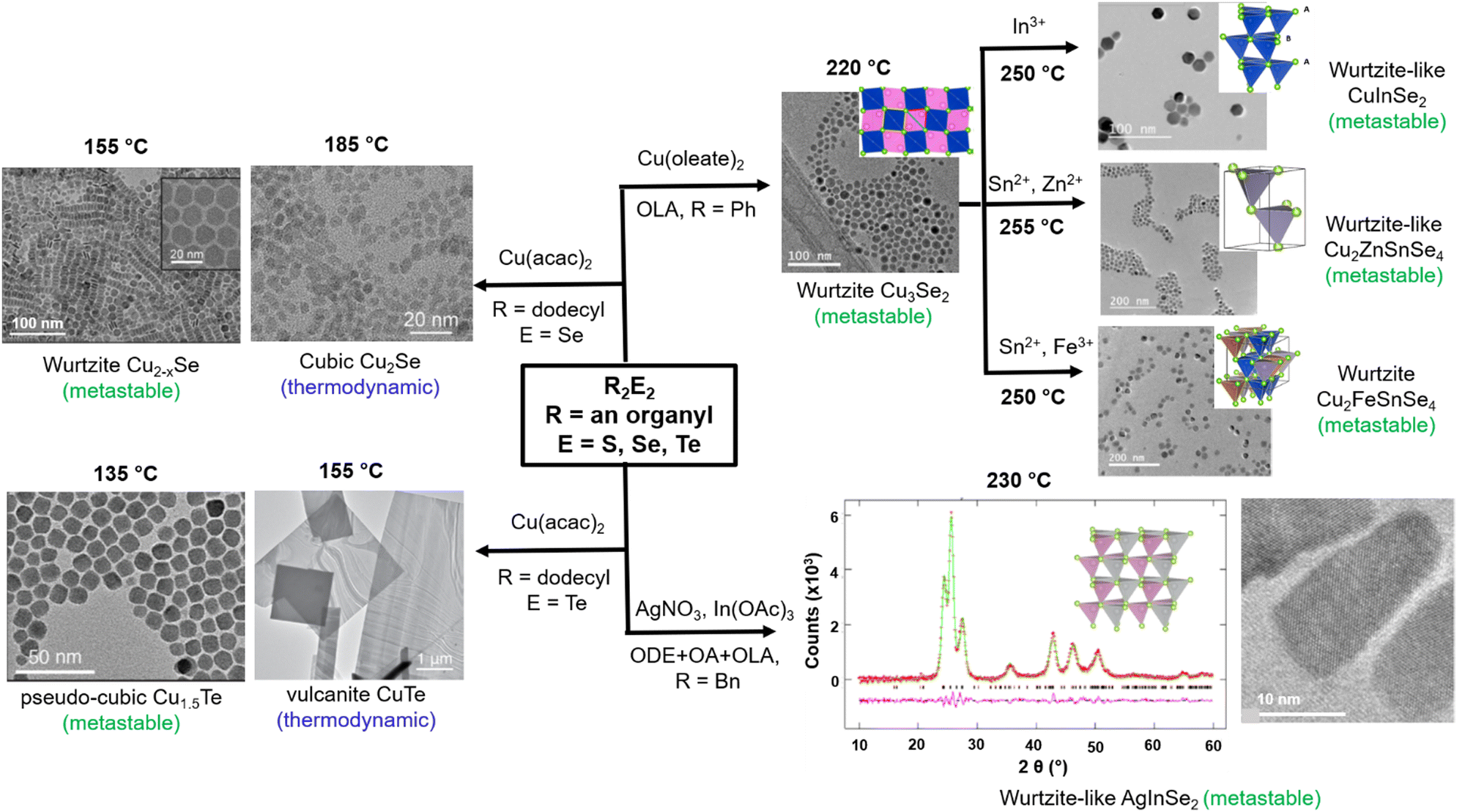 | ||
| Fig. 8 Isolation of metastable phases of binary and ternary metal chalcogenide NPs using R2E2 (R = an organyl; E = S, Se, Te) precursors. Images reproduced with permission from ref. 49–51 and 129–131. Copyright (2020) The Royal Society of Chemistry and (2018, 2019, 2020, 2021, 2021) American Chemical Society. | ||
The fact that the reaction of Ph2Se2 with Cu(oleate)2 in oleylamine yields preferentially the metastable Cu3Se2 intermediate which possesses a pseudo-hexagonal selenium sublattice that serves as a template for the ultimate wurtzite-like products has been exploited further to isolate many metastable phases. Thus, wurtzite-like metastable multinary Cu2FeSnSe4 and Cu2ZnSnSe4 have been isolated by in situ sequential cation exchange reactions between the intermediate Cu3Se2 NPs and metal precursors in solution.128,129 Besides the reactivity of the R2E2 reagent, the choice of surfactants employed also influences the course of the reaction and the nature of the final products. This is demonstrated by the reaction of Bz2Se2 with AgNO3 and In(OAc)3 in the presence of either oleic acid or oleylamine, which yielded thermodynamic chalcopyrite and metastable orthorhombic phases, respectively, of the ternary AgInSe2 NPs at 250 °C.130 The authors explained it on the basis of the tetragonal-to-orthorhombic phase transformation of the intermediate Ag2Se in the presence of oleylamine, which is then converted to the metastable orthorhombic phase of AgInSe2via fast In3+ cation exchange due to structural similarities between the two phases. In the absence of oleylamine, the Ag2Se intermediate is converted to the thermodynamic chalcopyrite structure of AgInSe2via kinetically slow nontopotactic conversion processes due to structural dissimilarities between the intermediate Ag2Se and chalcopyrite AgInSe2.
5. Conclusions and looking ahead
This feature article provides a concise and comparative overview on the chemistry and reactivity of three related families of organyl chalcogenides as convenient reagents for the ultra-mild synthesis of metal chalcogenide nanomaterials. These synthons not only transfer the chalcogenide anion (E2−) to a metal center under mild conditions but also allow their reactivity and properties to be modulated. Furthermore, their commercial availability, high solubility in common organic solvents and ability to react in a facile manner even with non-conventional metal reagents make them ideal precursors for scaled-up and controlled synthesis of nanometric metal chalcogenides.Unlike diorganyl dichalcogenides R2E2, the solution-phase utilization of diorganyl monochalcogenides has mostly been restricted to the silylated derivatives (R3Si)2E. Recent employment of nonsilylated diorganyl monochalcogenides R2E, which are less reactive than their silylated counterparts, has allowed the isolation and characterization of reactive molecular intermediates containing coinage metals during the synthesis of metal chalcogenides. An extension of this chemistry to other metals would help develop a broader understanding of the molecule-to-nanoparticle transformation and, therefore, achieve the synthesis of even compositionally complex materials with greater control. Recent work indicates that, by using non-silylated R2E, especially the ones that have a facile decomposition mechanism, it should also be possible to prepare a library of precursors that decompose at very low temperature to generate metal chalcogenide nanomaterials under ultra-mild conditions.43–46 These studies should be extended to the heterometallic complexes [MxM′y(R2E)z] containing two different metals or heteroleptic complexes [Mx(R2E)y(R2E′)z] with two different organyl chalcogenide ligands. These single source precursors with enhanced properties can overcome the problem of controlling the stoichiometry of the constituent elements, which often hinders the reproducibility in the synthesis of ternary metal chalcogenides. Reagents or precursors having comparable decomposition characteristics can also be a viable option for controlled synthesis of ternary phases, and despite some recent progress in it,63,92,93 better knowledge is required. A detailed computational and experimental study involving a systematic change of the R group to determine the variable reactivity in non-silylated R2E reagents would further allow us to control the reaction conditions, particularly to provide new synthetic pathways for kinetically-driven products. Previous studies on the R2E2 series have shown that the precursors possessing strong R–E bonds and therefore being less reactive are better suited for getting metastable phases.49 Non-silylated R2E, which are less reactive than silylated (R3Si)2E, have great potential in this regard.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support provided by the Université Claude Bernard Lyon 1 and the infrastructural support available at IRCELYON are thankfully acknowledged. The author also thanks his former PhD and master's students who worked in this field and whose names appear in the reference section.Notes and references
- X. Chen, J. Yang, T. Wu, L. Li, W. Luo, W. Jianga and L. Wang, Nanoscale, 2018, 10, 5130–15163 Search PubMed.
- C. Coughlan, M. Ibanez, O. Dobrozhan, A. Singh, A. Cabot and K. M. Ryan, Chem. Rev., 2017, 117, 5865–6109 CrossRef CAS PubMed.
- X. Cao, C. Tan, X. Zhang, W. Zhao and H. Zhang, Adv. Mater., 2016, 28, 6167–6196 CrossRef CAS PubMed.
- C. Han, Q. Sun, Z. Li and S. X. Dou, Adv. Energy Mater., 2016, 6, 1600498 CrossRef.
- L. Polavarapu, S. Mourdikoudis, I. Pastoriza-Santos and J. Perez-Juste, CrystEngComm, 2015, 17, 3727–3762 RSC.
- T. Heine, Acc. Chem. Res., 2015, 48, 65–72 CrossRef CAS PubMed.
- R. Gui, H. Jin, Z. Wang and L. Tan, Coord. Chem. Rev., 2015, 296, 91–124 CrossRef CAS.
- F. C. J. M. van Veggel, Chem. Mater., 2014, 26, 111–122 CrossRef CAS.
- S. R. Alvarado, Y. Guo, T. P. A. Ruberu, E. Tavasoli and J. Vela, Coord. Chem. Rev., 2014, 263–264, 182–196 CrossRef CAS.
- S. V. Kershaw, A. S. Susha and A. L. Rogach, Chem. Soc. Rev., 2013, 42, 3033–3087 RSC.
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulovic, Nat. Photonics, 2013, 7, 13–23 CrossRef CAS.
- J. Li and J. J. Zhu, Analyst, 2013, 138, 2506–2515 RSC.
- E. A. Weiss, Acc. Chem. Res., 2013, 46, 2607–2615 CrossRef CAS PubMed.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321–1324 CrossRef CAS PubMed.
- G. S. Li, D. Q. Zhang and J. C. Yu, Environ. Sci. Technol., 2009, 43, 7079–7085 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- M. A. Malik, M. Afzaal and P. O’Brien, Chem. Rev., 2010, 110, 4417–4446 CrossRef CAS PubMed.
- M. D. Khan, M. A. Malik and N. Revaprasadu, Coord. Chem. Rev., 2019, 388, 24–47 CrossRef CAS.
- V. Brune, M. Grosch, R. Weißing, F. Hartl, M. Frank, S. Mishra and S. Mathur, Dalton Trans., 2021, 50, 12365–12385 RSC.
- C. Glynn and C. O’Dwyer, Adv. Mater. Interfaces, 2017, 4, 1600610 CrossRef.
- C. Panda, P. W. Menezes and M. Driess, Angew. Chem., Int. Ed., 2018, 57, 11130–11139 CrossRef CAS PubMed.
- I. Bretos, R. Jimenez, J. Ricote and M. L. Calzada, Chem. Soc. Rev., 2018, 47, 291–308 RSC.
- S. Mishra and S. Daniele, Chem. – Eur. J., 2020, 26, 9292–9303 CrossRef CAS PubMed.
- Y. Guo, S. R. Alvarado, J. D. Barclay and J. Vela, ACS Nano, 2013, 7, 3616–3626 CrossRef CAS PubMed.
- R. L. Brutchey, Acc. Chem. Res., 2015, 48, 2918–2926 CrossRef CAS PubMed.
- J. M. Rhodes, C. A. Jones, L. B. Thal and J. E. Macdonald, Chem. Mater., 2017, 29, 8521–8530 CrossRef CAS.
- S. Dehnen, A. Eichhöfer and D. Fenske, Eur. J. Inorg. Chem., 2002, 279–2317 CrossRef.
- J. F. Corrigan, O. Fuhr and D. Fenske, Adv. Mater., 2009, 21, 1867–1871 CrossRef CAS.
- O. Fuhr, S. Dehnen and D. Fenske, Chem. Soc. Rev., 2013, 42, 1871–1906 RSC.
- D. G. Macdonald and J. F. Corrigan, Philos. Trans. R. Soc., A, 2010, 368, 1455–1472 CrossRef CAS PubMed.
- Y. P. Chang, W. Levason and G. Reid, Dalton Trans., 2016, 45, 18393–18416 RSC.
- W. Levason, G. Reid and W. Zhang, Dalton Trans., 2011, 40, 8491–8506 RSC.
- N. D. Boscher, C. J. Carmalt and I. P. Parkin, J. Mater. Chem., 2006, 16, 122–127 RSC.
- N. D. Boscher, C. J. Carmalt, R. G. Palgrave and I. P. Parkin, Thin Solid Films, 2008, 516, 4750–4757 CrossRef CAS.
- P. I. Kuznetsov, V. O. Yapaskurt, B. S. Shchamkhalova, V. D. Shcherbakov, G. G. Yakushcheva, V. A. Luzanova and V. A. Jitov, J. Cryst. Growth, 2016, 455, 122–128 CrossRef CAS.
- B. J. Choi, S. Choi, Y. C. Shin, K. M. Kim, C. S. Hwang, Y. J. Kim, Y. J. Son and S. K. Hong, Chem. Mater., 2007, 19, 4387–4389 CrossRef CAS.
- S. L. Benjamin, Y. Chang, C. Gurnani, A. L. Hector, M. Huggon, W. Levason and G. Reid, Dalton Trans., 2014, 43, 16640–16648 RSC.
- S. L. Benjamin, C. H. (Kees) de Groot, C. Gurnani, A. L. Hector, R. Huang, E. Koukharenko, W. Levason and G. Reid, J. Mater. Chem. A, 2014, 2, 4865–4869 RSC.
- S. L. Benjamin, C. H. (Kees) de Groot, C. Gurnani, S. L. Hawken, A. L. Hector, R. Huang, M. Jura, W. Levason, E. Reid, G. Reid, S. P. Richards and G. B. G. Stenning, J. Mater. Chem. C, 2018, 6, 7734–7739 RSC.
- K. George, C. H. (Kees) de Groot, C. Gurnani, A. L. Hector, R. Huang, M. Jura, W. Levason and G. Reid, Chem. Mater., 2013, 25, 1829–1836 CrossRef CAS.
- M. E. Norako, M. J. Greaney and R. L. Brutchey, J. Am. Chem. Soc., 2012, 134, 23–26 CrossRef CAS PubMed.
- S. Gahlot, B. Purohit, E. Jeanneau and S. Mishra, Chem. – Eur. J., 2021, 27, 10826–10832 CrossRef CAS PubMed.
- S. Mishra, D. Du, E. Jeanneau, F. Dappozze, C. Guillard, J. Zhang and S. Daniele, Chem. – Asian J., 2016, 11, 1658–1663 CrossRef CAS PubMed.
- S. Gahlot, E. Jeanneau, F. Dappozze, C. Guillard and S. Mishra, Dalton Trans., 2018, 47, 8897–8905 RSC.
- S. Gahlot, E. Jeanneau, D. Singh, P. K. Panda, Y. K. Mishra, R. Ahuja, G. Ledoux and S. Mishra, Inorg. Chem., 2020, 59, 7727–7738 CrossRef CAS PubMed.
- S. Gahlot, F. Dappozze, D. Singh, R. Ahuja, L. Cardenas, L. Burel, D. Amans, C. Guillard and S. Mishra, Dalton Trans., 2020, 49, 3580–3591 RSC.
- S. Mishra and S. Gahlot, Nanomaterials via Single-Source Precursors: Synthesis, Processing and Applications, ed. A. W. Apblett, A. R. Barron and A. F. Hepp, Elsevier, 2022, ch. 6, pp. 201–218 Search PubMed.
- (a) B. A. Tappan, G. Barim, J. C. Kwok and R. L. Brutchey, Chem. Mater., 2018, 30, 5704–5713 CrossRef CAS; (b) M. E. Norako and R. L. Brutchey, Chem. Mater., 2010, 22, 1613–1615 CrossRef CAS.
- E. A. Hernández-Pagán, E. H. Robinson, A. D. La Croix and J. E. Macdonald, Chem. Mater., 2019, 31, 4619–4624 CrossRef.
- E. H. Robinson, K. M. Dwyer, A. C. Koziel, A. Y. Nuriye and J. E. Macdonald, Nanoscale, 2020, 12, 23036–23041 RSC.
- S. Schulz, Coord. Chem. Rev., 2015, 297–298, 49–76 CrossRef CAS.
- N. L. Pickett, D. F. Foster, N. Maung and D. J. Cole-Hamilton, J. Mater. Chem., 1999, 9, 3005–3014 RSC.
- C. A. Class, M. Liu, A. G. Vandeputte and W. H. Green, Phys. Chem. Chem. Phys., 2016, 18, 21651–21658 RSC.
- G. A. Seisenbaeva and V. G. Kessler, Nanoscale, 2014, 6, 6229–6244 RSC.
- S. Mishra and S. Daniele, Chem. Rev., 2015, 115, 8379–8448 CrossRef CAS PubMed.
- S. Mishra, S. Daniele and L. G. Hubert-Pfalzgraf, Chem. Soc. Rev., 2007, 36, 1770–1787 RSC.
- S. L. Benjamin, C. H. (Kees) de Groot, C. Gurnani, A. L. Hector, R. Huang, K. Ignatyev, W. Levason, S. J. Pearce, F. Thomas and G. Reid, Chem. Mater., 2013, 25, 4719–4724 CrossRef CAS PubMed.
- Y. Chang, A. L. Hector, W. Levason, G. Reid and J. Whittam, Dalton Trans., 2018, 47, 2406–2414 RSC.
- C. Gurnani, S. L. Hawken, A. L. Hector, R. Huang, M. Jura, W. Levason, J. Perkins, G. Reid and G. B. G. Stenningd, Dalton Trans., 2018, 47, 2628–2637 RSC.
- S. Mishra, E. Jeanneau and S. Daniele, Polyhedron, 2010, 29, 500–506 CrossRef CAS.
- K. T. Higa and K. A. Fallis, J. Cluster Sci., 2002, 13, 533–541 CrossRef CAS.
- S. Schulz, S. Heimann, J. Friedrich, M. Engenhorst, G. Schierning and W. Assen-macher, Chem. Mater., 2012, 24, 2228–2234 CrossRef CAS.
- S. Heimann, W. Assenmacher, O. Prymak and S. Schulz, Eur. J. Inorg. Chem., 2015, 2407–2415 CrossRef CAS.
- G. Bendt, A. Weber, S. Heimann, W. Assenmacher, O. Prymak and S. Schulz, Dalton Trans., 2015, 44, 14272–14280 RSC.
- S. Heimann, S. Schulz, J. Schaumann, A. Mudring, J. Stotzel and G. Schierning, J. Mater. Chem. C, 2015, 3, 10375–10380 RSC.
- J. Schaumann, M. Loor, D. Unal, A. Mudring, S. Heimann, U. Hagemann, S. Schulz, F. Maculewicz and G. Schierning, Dalton Trans., 2017, 46, 656–668 RSC.
- M. Loor, G. Bendt, U. Hagemann, C. Wolper, W. Assenmacher and S. Schulz, Dalton Trans., 2016, 45, 15326–15335 RSC.
- G. Gupta and J. Kim, J. Chem. Soc., Dalton Trans., 2013, 42, 8209–8211 RSC.
- G. Bendt, S. Gassa, F. Rieger, C. Jooss and S. Schulz, J. Cryst. Growth, 2018, 490, 77–83 CrossRef CAS.
- K. T. Higa and K. A. Fallis, J. Cluster Sci., 2002, 13, 533–541 CrossRef CAS.
- M. Gomez, C. Hernandez-Prieto, A. Martín, M. Mena and C. Santamaría, Inorg. Chem., 2016, 55, 3815–3821 CrossRef CAS PubMed.
- J. P. Eußner, R. O. Kusche and S. Dehnen, Chem. – Eur. J., 2015, 21, 12376–12388 CrossRef PubMed.
- G. Bendt, A. Weber, S. Heimann, W. Assenmacher, O. Prymak and S. Schulz, Dalton Trans., 2015, 44, 14272–14280 RSC.
- E. Dornsiepen and S. Dehnen, Dalton Trans., 2019, 48, 3671–3675 RSC.
- D. Fuhrmann and H. Krautscheid, Z. Anorg. Allg. Chem., 2022 DOI:10.1002/zaac.202200099.
- M. A. Fard, F. Weigend and J. F. Corrigan, Chem. Commun., 2015, 51, 8361–8364 RSC.
- M. W. DeGroot and J. F. Corrigan, Organometallics, 2005, 24, 3378–3385 CrossRef CAS.
- M. A. Fard, T. I. Levchenko, C. Cadogan, W. J. Humenny and J. F. Corrigan, Chem. – Eur. J., 2016, 22, 4543–4550 CrossRef PubMed.
- H. Lu and R. L. Brutchey, Chem. Mater., 2017, 29, 1396–1403 CrossRef CAS.
- M. A. Hine and G. D. Scholes, Adv. Mater., 2003, 15, 1844–1849 CrossRef.
- J. Tang, L. Brzozowski, D. A. R. Barkhouse, X. Wang, R. Debnath, R. Wolowiec, E. Palmiano, L. Levina, A. G. Pattantyus-Abraham and D. Jamakosmanovic, ACS Nano, 2010, 4, 869–878 CrossRef CAS PubMed.
- N. Zhao, T. P. Osedach, L.-Y. Chang, S. M. Geyer, D. Wanger, M. T. Binda, A. C. Arango, M. G. Bawendi and V. Bulovic, ACS Nano, 2010, 4, 3743–3752 CrossRef CAS PubMed.
- N. J. Thompson, M. W. B. Wilson, D. N. Congreve, P. R. Brown, J. M. Scherer, T. S. Bischof, M. Wu, N. Geva, M. Welborn, T. V. Voorhis, V. Bulovic, M. G. Bawendi and M. A. Baldo, Nat. Mater., 2014, 13, 1039–1043 CrossRef CAS PubMed.
- A. Stavrinadis, D. So and G. Konstantatos, J. Phys. Chem. C, 2016, 120, 20315–20322 CrossRef CAS.
- J. Zhang, J. Gao, E. M. Miller, J. M. Luther and M. C. Beard, ACS Nano, 2014, 8, 614–622 CrossRef CAS PubMed.
- Y. Zhang, G. Wu, C. Ding, F. Liu, Y. Yao, Y. Zhou, C. Wu, N. Nakazawa, Q. Huang, T. Toyoda, R. Wang, S. Hayase, Z. Zou and Q. Shen, J. Phys. Chem. Lett., 2018, 9, 3598–3603 CrossRef CAS PubMed.
- J. Akhtar, M. A. Malik, P. O’Brien, K. G. U. Wijayantha, R. Dharmadasa, S. J. O. Hardman, D. M. Graham, B. F. Spencer, S. K. Stubbs, W. R. Flavell, D. J. Binks, F. Sirotti, M. El Kazzi and M. Silly, J. Mater. Chem., 2010, 20, 2336–2344 RSC.
- H. Zhang, B. H. Savitzky, J. Yang, J. T. Newman, K. A. Perez, B.-R. Hyun, L. F. Kourkoutis, T. Hanrath and F. W. Wise, Chem. Mater., 2016, 28, 127–134 CrossRef CAS.
- A. Shrestha, M. Batmunkh, A. Tricoli, S. Z. Qiao and S. Dai, Angew. Chem., Int. Ed., 2019, 58, 5202–5224 CrossRef CAS PubMed.
- A. Antanovich, A. Prudnikau and M. Artemyev, J. Phys. Chem. C, 2014, 118, 21104–21109 CrossRef CAS.
- D. K. Smith, J. M. Luther, O. E. Semonin, A. J. Nozik and M. C. Beard, ACS Nano, 2011, 5, 183–190 CrossRef CAS PubMed.
- J. Akhtar, M. Afzaal, M. Banski, A. Podhorodecki, M. Syperek, J. Misiewicz, U. Bangert, S. J. O. Hardman, D. M. Graham and W. R. Flavell, J. Am. Chem. Soc., 2011, 133, 5602–5609 CrossRef CAS PubMed.
- G. Shen, M. Chen and P. Guyot-Sionnest, J. Phys. Chem. Lett., 2017, 8, 2224–2228 CrossRef CAS PubMed.
- P. Glaser, O. Stewart, Jr., R. Atif, D. R. C. Asuigui, J. Swanson, A. J. Biacchi, A. R. H. Walker, G. Morrison, H.-C. zur Loye and S. L. Stoll, Angew. Chem., Int. Ed., 2021, 60, 23134–23141 CrossRef CAS PubMed.
- V. A. Ermakov, J. M. C. S. Filho, L. G. Bonato, N. V. V. Mogili, F. E. Montoro, F. Iikawa, A. F. Nogueira, C. L. Cesar, E. Jimenez-Villar and F. C. Marques, ACS Omega, 2018, 3, 2027–2032 CrossRef CAS PubMed.
- (a) S. Schulz, S. Heimann, K. Kaiser, O. Prymak, W. Assenmacher, J. T. Bruggemann, B. Mallick and A. V. Mudring, Inorg. Chem., 2013, 52, 14326–14333 CrossRef CAS PubMed; (b) J. M. Hwang, J. H. Lee, H.-S. Kim, C. W. Park, D. Yoo, B. K. Park, C. G. Kim and T.-M. Chung, Polyhedron, 2020, 176, 14289–114294 CrossRef.
- M. Rusek, G. Bendt, C. Wölper and S. Schulz, Eur. J. Inorg. Chem., 2016, 3673–3679 CrossRef CAS.
- B. Zhou, M. Li, Y. Wu, C. Yang, W. H. Zhang and C. Li, Chem. – Eur. J., 2015, 21, 11143–11151 CrossRef CAS PubMed.
- P. M. Allen and M. G. Bawendi, J. Am. Chem. Soc., 2008, 130, 9240–9241 CrossRef CAS PubMed.
- S. Jung, J.-H. Cha and D.-Y. Jung, Thin Solid Films, 2016, 603, 243–248 CrossRef CAS.
- M. Loor, G. Bendt, J. Schaumann, U. Hagemann, M. Heidelmann, C. Wolper and S. Schulz, Z. Anorg. Allg. Chem., 2017, 643, 60–68 CrossRef CAS.
- S. Salloum, G. Bendt, M. Heidelmann, K. Loza, S. Bayesteh, M. S. Izadi, P. Kawulok, R. He, H. Schlorb, N. Perez, H. Reith, K. Nielsch, G. Schierning and S. Schulz, ChemistryOpen, 2021, 10, 189–198 CrossRef CAS PubMed.
- J. Guschlbauer and J. Sundermeyer, ChemistryOpen, 2021, 10, 92–96 CrossRef CAS PubMed.
- M. Loor, S. Salloum, P. Kawulok, S. Izadi, G. Bendt, J. Guschlbauer, J. Sundermeyer, N. Perez, K. Nielsch, G. Schierning and S. Schulz, Inorg. Chem., 2020, 59, 3428–3436 CrossRef CAS PubMed.
- E. A. Turner, H. Ro1sner, D. Fenske, Y. Huang and J. F. Corrigan, J. Phys. Chem. B, 2006, 110, 16261–16269 CrossRef CAS PubMed.
- E. A. Turner, H. Rosner, Y. Huang and J. F. Corrigan, J. Phys. Chem. C, 2007, 111, 7319–7329 CrossRef CAS.
- A. Wolf, T. Hrtling, D. Hinrichs and D. Dorfs, ChemPhysChem, 2016, 17, 717–723 CrossRef CAS PubMed.
- J. M. Hodges, K. Kletetschka, J. L. Fenton, C. G. Read and R. E. Schaak, Angew. Chem., Int. Ed., 2015, 54, 8669–8672 CrossRef CAS PubMed.
- E. E. Foos, R. M. Stroud and A. D. Berry, Nano Lett., 2001, 1, 693–695 CrossRef CAS.
- J. L. Fenton, A. M. Fagan and R. E. Schaak, Eur. J. Inorg. Chem., 2019, 3490–3493 CrossRef CAS.
- J. Park, H. Zheng, Y.-W. Jun and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 13943–13945 CrossRef CAS PubMed.
- C. C. Lin, S. J. Tana and J. Vela, J. Mater. Chem. A, 2017, 5, 20351–20358 RSC.
- S. Schlecht, M. Budde and L. Kienle, Inorg. Chem., 2002, 41, 6001–6005 CrossRef CAS PubMed.
- P. J. McKarns, T. S. Lewkebandara, G. P. A. Yap, L. M. Liable-Sands, A. L. Rheingold and C. H. Winter, Inorg. Chem., 1998, 37, 418–424 CrossRef CAS PubMed.
- A. K. FAzlur-Rahman and J. G. Verkade, Inorg. Chem., 1992, 31, 2064–2069 CrossRef CAS.
- E. A. Turner, H. Rosner, Y. Huang and J. F. Corrigan, J. Cluster Sci., 2007, 18, 764–771 CrossRef CAS.
- E. A. Turner, H. Rosner, Y. Huang and J. F. Corrigan, Can. J. Chem., 2007, 85, 747–755 CrossRef CAS.
- D. H. Webber and R. L. Brutchey, Chem. Commun., 2009, 5701–5703 RSC.
- D. H. Webber and R. L. Brutchey, Inorg. Chem., 2011, 50, 723–725 CrossRef CAS PubMed.
- D.-J. Xue, J. Tan, J.-S. Hu, W. Hu, Y.-G. Guo and L.-J. Wan, Adv. Mater., 2012, 24, 4528–4533 CrossRef CAS PubMed.
- M. A. Franzman, C. W. Schlenker, M. E. Thompson and R. L. Brutchey, J. Am. Chem. Soc., 2010, 132, 4060–4061 CrossRef CAS PubMed.
- J. J. Buckley, F. A. Rabuffetti, H. L. Hinton and R. L. Brutchey, Chem. Mater., 2012, 24, 3514–3516 CrossRef CAS.
- M. A. Franzman and R. L. Brutchey, Chem. Mater., 2009, 21, 1790–1792 CrossRef CAS.
- M. E. Norako, M. A. Franzman and R. L. Brutchey, Chem. Mater., 2009, 21, 4299–4304 CrossRef CAS.
- D. Sun, Y. Xiong, Y. Sun, I. Dabo and R. E. Schaak, Chem. Mater., 2017, 29, 1095–1098 CrossRef CAS.
- Y.-K. Hong, Y. Park, H. Kang, H. Son and D.-H. Ha, Dalton Trans., 2021, 50, 2192–2199 RSC.
- W. Li, A. Shavel, R. Guzman, J. Rubio-Garcia, C. Flox, J. Fan, D. Cadavid, M. Ibanez, J. Arbiol, J. R. Morantea and A. Cabot, Chem. Commun., 2011, 47, 10332–10334 RSC.
- B. A. Tappan, W. Chu, M. Mecklenburg, O. V. Prezhdo and R. L. Brutchey, ACS Nano, 2021, 15, 13463–13474 CrossRef CAS PubMed.
- B. A. Tappan, K. D. Crans and R. L. Brutchey, Inorg. Chem., 2021, 60, 17178–17185 CrossRef CAS PubMed.
- B. A. Tappan, K. D. Crans and R. L. Brutchey, Chem. Mater., 2020, 32, 2935–2945 CrossRef CAS.
- W. Wang, W. Feng, Q. Li, Y. Zhao, D. Zhao, Z. Xia, W. Wang, S. Zhang, X. Zheng and Z. Jing, Cryst. Growth Des., 2019, 19, 1226–1232 CrossRef CAS.
- P. D. Antunez, D. H. Webber and R. L. Brutchey, Chem. Mater., 2013, 25, 2385–2387 CrossRef CAS.
- J. Q. Geisenhoff, A. K. Tamura and A. M. Schimpf, Chem. Commun., 2019, 55, 8856–8859 RSC.
- W. Jung, S. Lee, D. Yoo, S. Jeong, P. Miro, A. Kuc, T. Heine and J. Cheon, J. Am. Chem. Soc., 2015, 137, 7266–7269 CrossRef CAS PubMed.
- Y. Sun, K. Fujisawa, Z. Lin, Y. Lei, J. S. Mondschein, M. Terrones and R. E. Schaak, J. Am. Chem. Soc., 2017, 139, 11096–11105 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |