
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereoselective access to substituted oxetanes via hydrosilylation–iodocyclisation of homopropargylic alcohols†
Dean D.
Roberts
and
Mark G.
McLaughlin
 *
*
Department of Chemistry, University of Lancaster, Bailrigg, Lancashire LA14YB, UK. E-mail: m.mclaughlin3@lancaster.ac.uk
First published on 30th June 2022
Abstract
The regio and stereoselective hydrosilylation of a variety of homopropargylic alcohols and their derivatives is described. The reaction is tolerant to a variety of sterically and electronically varied substrates, affording only the E-vinyl silane as a sole regioisomer. The application of the resultant vinyl silanes towards the diastereoselective synthesis of tetrasubstituted oxetanes is demonstrated.
The use of organosilanes remains central to modern organic chemistry, in no small part to the plethora of transformations which these groups are amenable to. In particular, organosilicon compounds find utility as nucleophilic coupling partners in the Hiyama cross-coupling and as masked hydroxyl groups in the Fleming–Tamao oxidation.1,2 In comparison to other classes of organometalloid compounds, organosilanes are relatively non-toxic, and display good levels of air and moisture stability, aiding ease of manipulation. Development of methods to access organosilanes which are operationally simple and robust has therefore remained a key goal within synthetic chemistry. Hydrosilylation of unsaturated bonds is one such method which has many well-established successes. This is a particularly attractive process as it affords the product in 100% atom economy, minimising the amount of waste generated throughout the transformation.3 Catalytic hydrosilylation processes generally employ late transition metals, with Pt catalysts being the most frequently utilised after the discovery of hexachloroplatinic acid in the 1950s.4–6 Whilst these Pt based catalysts benefit from high turnover numbers/frequencies, introduction of coordinating functional groups has often led to catalyst poisoning, either leading to deactivation or poor regioselectivity and stereoselectivity of the resultant hydrosilylation.7–9 Recently we disclosed that through judicious ligand choice this inhibition can be overcome, allowing for the hydrosilylation of propargylic amines.10
The issue of directed metalation leading to poor regioselectivity is not unique to Pt catalysts however, with thioethers being shown to direct the hydrosilylation of alkynes using both Co and Ru catalysts (Fig. 1).11,12 The affinity of transition metals for Lewis basic functional groups has been utilised as a method for directing hydrosilylation towards a specific regioisomer through chelate control. This approach was utilised by Van Vranken and coworkers in their Pt catalysed hydrosilylation of homoallylic thioethers.13 Recently, Ding and co-workers screened a range of directing groups in their study of the directed Ir catalysed hydrosilylation of alkynes. They found that whilst nitrogen- and sulfur-containing functional groups almost completely inhibited the reaction, oxygen containing functionalities, such as alcohols, esters and silyl, were able to competently direct the hydrosilylation.14 This corroborates earlier findings which suggest that oxygen containing groups are effective at directing the Ir-catalysed hydrosilylation of terminal alkynes, with the α-isomer preferentially formed in a 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in the case of homopropargylic alcohols.15 This is in agreement with earlier examples of hydroxyl directed hydrosilylations by Chang and Na.16 As previous work in the group has demonstrated the cyclisation of vinyl silanes bearing a tethered nucleophile towards the synthesis of aziridines, we wished to explore the synthesis of oxetanes using similar methodology affording oxetanes with an ambiphillic α-iodosilane centre, a functionality which has shown to have unique applications as an electrophile in enantioselective cross-couplings, as reported by Oestreich.17 Whilst halocyclization reactions have been reported previously, they either rely on stoichiometric amounts of strong bases18 or in the case of silyl–olefins, elevated reaction temperatures as reported by Rousseau.19 As well as being high value synthetic intermediates, oxetanes are privileged fragments in medicinal chemistry and are frequently utilised as bioisosteres.20 In addition to this, the oxetane motif is found in a variety of natural products. Existing methods to access these scaffolds typically involve the use of stoichiometric amounts of strong base to realise an intramolecular Williamson ether synthesis, or require the use of complex photochemical reaction set-ups to carry out the Paterno–Buchi reaction (Fig. 2).21 As noted by Gouverneur et al., the cyclisation of allylic silanes affords the cyclised product, where the stereochemical relationship of the exocyclic functionalities is dependent on the alkene geometry.22
:1 ratio in the case of homopropargylic alcohols.15 This is in agreement with earlier examples of hydroxyl directed hydrosilylations by Chang and Na.16 As previous work in the group has demonstrated the cyclisation of vinyl silanes bearing a tethered nucleophile towards the synthesis of aziridines, we wished to explore the synthesis of oxetanes using similar methodology affording oxetanes with an ambiphillic α-iodosilane centre, a functionality which has shown to have unique applications as an electrophile in enantioselective cross-couplings, as reported by Oestreich.17 Whilst halocyclization reactions have been reported previously, they either rely on stoichiometric amounts of strong bases18 or in the case of silyl–olefins, elevated reaction temperatures as reported by Rousseau.19 As well as being high value synthetic intermediates, oxetanes are privileged fragments in medicinal chemistry and are frequently utilised as bioisosteres.20 In addition to this, the oxetane motif is found in a variety of natural products. Existing methods to access these scaffolds typically involve the use of stoichiometric amounts of strong base to realise an intramolecular Williamson ether synthesis, or require the use of complex photochemical reaction set-ups to carry out the Paterno–Buchi reaction (Fig. 2).21 As noted by Gouverneur et al., the cyclisation of allylic silanes affords the cyclised product, where the stereochemical relationship of the exocyclic functionalities is dependent on the alkene geometry.22
 | ||
| Fig. 1 Applications of co-ordinating functional groups as directing groups in alkyne hydrosilylation. | ||
 | ||
| Fig. 2 Cyclisation of unsaturated silanes towards the synthesis of heterocycles. | ||
As the alkenes in Gouverneur's work were a mixture of isomers, this afforded the cyclised product as a mixture of diastereomers. We reasoned this drawback could be overcome if the silane was accessed via the hydrosilylation chemistry previously published by our group.23 We therefore aimed to develop a synthetic method for the hydrosilylation of homopropargylic alcohols which overcomes the previously noted directing group abilities of the hydroxyl group, and affords the corresponding vinyl silane as a sole regio and stereoisomer. We subsequently aimed to explore the cyclisation of the resultant vinyl silanes into oxetanes through an electrophilic activation strategy by taking advantage of the well-established β-silicon effect.24
We began our investigation using model substrate 1a employing 10 mol% of PtCl2 (Scheme 1). Whilst we were pleased to observe consumption of the starting alkyne, 1H NMR analysis of the crude reaction mixture indicated the presence of the branched alkene arising from chelate directed hydrosilylation. Gratifyingly, when PtCl2:XPhos was employed, this regioselectivity loss could be mitigated, with 100% conversion to branched E-vinyl silane observed. Reduction in the catalyst loading had little effect on the reaction, with 1 mol% with respect to platinum affording the product with complete control. Upon a survey of other phosphine ligands, we found that XPhos was unique in its ability to achieve this regioselectivity. Further screens of solvents and temperatures were likewise unable to match the efficacy of entry 4, likely due to the poor solubility of the platinum complex.
 | ||
| Scheme 1 Optimisation study. | ||
With an optimised catalyst system and reaction conditions now in hand, we turned our attention to exploring the scope of alkynes which were amenable to the reaction (Scheme 2). Beginning our study with secondary homopropargylic alcohols, we were delighted to observe that the reaction was tolerant to electron-deficient aromatics, with halides tolerated in different positions around the ring (2b, 2c) with negligible impact on the yield of the reaction. The reaction was equally successful with more electron-rich aromatics (2d, 2i.) It is noteworthy that substrates bearing heterocyclic scaffolds were also amenable to hydrosilylation, with thiophenyl silane (2o) accessed in 70% yield, though pyridyl groups appear to be poorly tolerated by the reaction (2t). Substrates bearing bulky cyclohexyl and tert-butyl groups were also suitable substrates for the reaction, again with only the E-vinyl silane formed in excellent yields. We then turned our attention to exploring the scope of the reaction with respect to more sterically encumbered tertiary alcohols. We were pleased to observe that the optimised conditions for the secondary al-cohols were equally as successful in the hydrosilylation of these substrates, with the diphenyl and fluorenyl alcohols (2r, 2s) afforded in excellent yields. Symmetrical alkyl substrates were equally suitable substrates (2q). We finally wanted to probe the efficacy of the reaction when other hydrosilanes were used in place of the previously used dimethylbenzyl silane. To this end, we screened a variety of alkyl, aryl and alkoxy hydrosilanes under the reaction conditions, with 2 alkynes of varying sterics and electronics. In the case of both alkynes, HSiMe2Ph, HSi(OEt)3 and HSiEt3 were all viable hydrosilylating reagents, although no reaction was observed in the case of HSi(iPr)3.
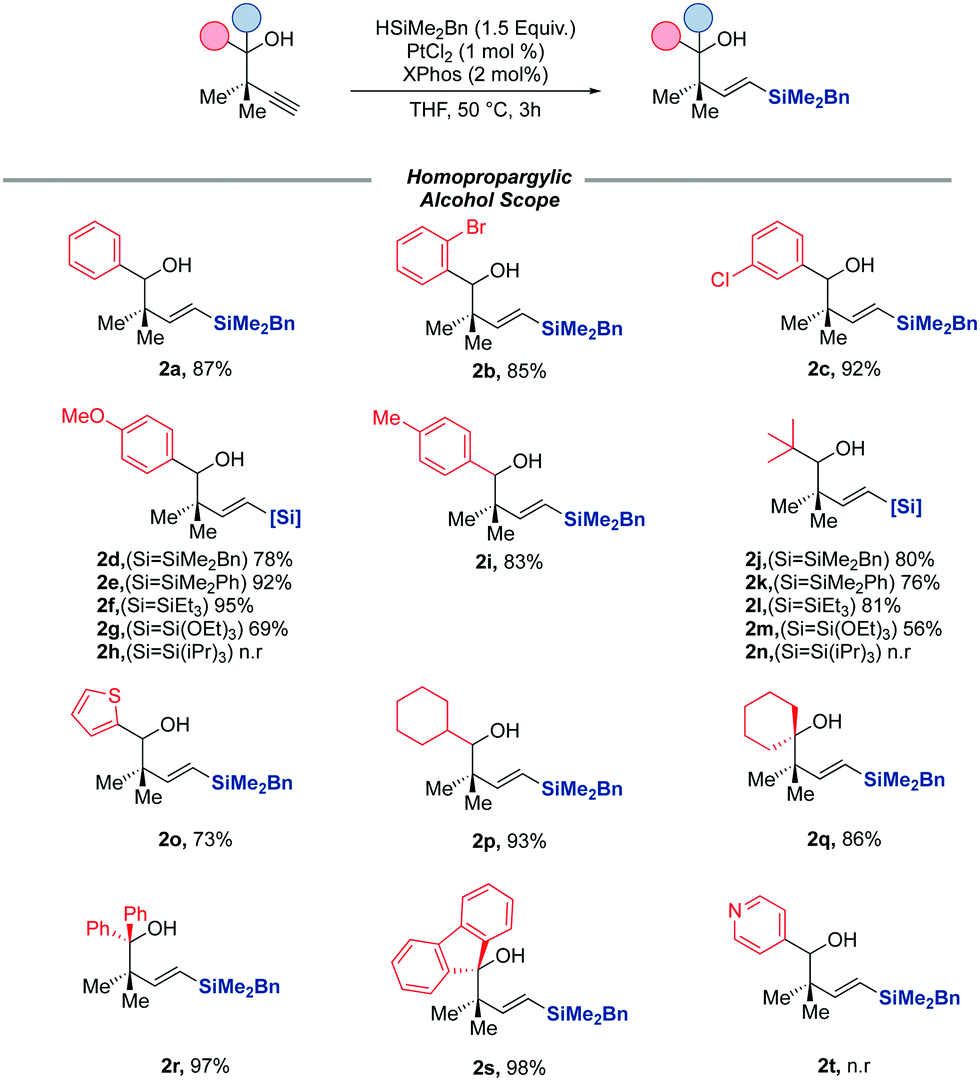 | ||
| Scheme 2 Homopropargylic alcohol scope. | ||
With a range of homoallylic alcohols accessed by this methodology, we were interested to probe whether the methodology could be extended to other homopropargylic heteroatomic functionalities, particularly those which could introduce further points of contact with the catalyst system. We were initially concerned that the introduction of new sites for metal–substrate interaction could hinder the selectivity of the reaction, through chelate directed metalation (Scheme 3). We began by carrying out the hydrosilylation of homopropargylic ethers. Benzyl ethers of varying electronics and substitution patterns were viable substrates (4a, 4b). We then examined the hydrosilylation of homopropargylic esters, and were delighted to observe that our concerns of chelate directed metalation were misplaced, with the corresponding vinyl silane formed as a sole regio and stereoisomer (4c, 4d). Finally, we wanted to explore the hydrosilylation of nitrogen containing functionalities. To this end, we prepared amide 3e and subjected to the reaction conditions, affording 4e in 93% yield. Carrying out the hydrosilylation of the free carboxylic acid 3f resulted in the formation of a complex inseparable mixture of products.
 | ||
| Scheme 3 Further functionalisation. | ||
Using the conditions previously reported by our group in the previous halocyclisations of vinyl silanes, we attempted the cyclisation of 2a. Whilst we observed complete consumption of the vinyl silane, and formation of the corresponding oxetane, we also observed formation of the corresponding vinyl iodide, formed either via competing desilylation or succinimide mediated ring-opening of the oxetane. We therefore reasoned that carrying out the reaction under basic conditions would supress the desilylation pathways. To this end, we carried out the reaction again, this time in the presence of NaHCO3, which afforded oxetane 5a in 87% yield, with no observable vinyl iodide formation. As noted earlier, the alkene geometry has shown to have a profound effect in the stereochemical outcome of the cyclisation of unsaturated silanes. As the starting alkene was geometrically pure, we had expected high levels of diastereoselectivity to be observed in the product, and we were delighted to observe formation of the corresponding oxetane in high levels of diastereoselectivity. NOESY studies showed that the substituents in the 1 and 3 positions were mutually trans (see ESI†) Using these conditions, we were able to access a range of oxetanes bearing aliphatic, aromatic and heteroaromatic substituents in good to excellent yields (Scheme 4). Attempted cyclisation of more sterically encumbered alcohols 2r and 2s resulted in low conversion observed in the 1H NMR of the crude reaction mixture, whilst heating the reaction mixture only increased the amount of vinyl iodide in the reaction mixture.
 | ||
| Scheme 4 Oxetanes accessed via halocyclisation. | ||
In addition to applications in the synthesis of oxetanes, we also wished to demonstrate the applicability of products of this work. Hiyama coupling to afford non-silylated homoallylic alcohols, compounds which are versatile intermediates in their own right.25 Using previously reported conditions for the coupling of vinyldimethylbenzyl silanes, we carried out the palladium–catalysed cross–coupling of 2j with iodobenzene.26 and obtained the cross-coupled product 6a in 89% yield, with no loss of geometric purity of the alkene (Scheme 5).
 | ||
| Scheme 5 Hiyama cross-coupling of homoallylic alcohol. | ||
In conclusion, we had developed the first regioselective and stereoselective hydrometallation of homopropargyl heteroa-tom containing scaffolds using a platinum/XPhos catalyst system at 1 mol% catalyst loading. The reaction is tolerant to a variety of electronically varied alkynes, with the vinyl silanes affording in good to excellent yields as a sole regio and stereoisomer. Additionally, the reaction can be scaled to the multi-mmol scale with negligible effect on reaction efficacy. A variety of homopropargylic scaffolds are amenable to the reaction, including those which have previously acted as directing groups in hydrosilylation chemistry such as ethers, esters and amides. The application of the compounds towards the synthesis of oxetanes has also been disclosed, with the reaction proceeding through electrophilic activation of the vinyl silane. The resultant cyclisation proceeds with high levels of diastereoselectivity, with the groups in the 1 and 3 positions of the ring being mutually trans to one another.
We thank the Royal Society of Chemistry for the award of a Research Enablement Grant. DDR thanks Lancaster University for a studentship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918–920 CrossRef CAS.
- K. Tamao, N. Ishida, T. Tanaka and M. Kumada, Organometallics, 1983, 2, 1694–1696 CrossRef CAS.
- B. M. Trost and Z. T. Ball, Synthesis, 2005, 853–887 CrossRef CAS.
- J. L. Speier, J. A. Webster and G. H. Barnes, J. Am. Chem. Soc., 1957, 79, 974–979 CrossRef CAS.
- J. C. Saam and J. L. Speier, J. Am. Chem. Soc., 1958, 80, 4104–4106 CrossRef CAS.
- M. P. Sibi, Encyclopedia of Reagents for Organic Synthesis, 2001 DOI:10.1002/047084289X.rh038.
- K. Kishi, T. Ishimaru, M. Ozono, I. Tomita and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 804–809 CrossRef CAS.
- J. Saam and J. Speier, J. Org. Chem., 1959, 24, 119–120 CrossRef CAS.
- A. Chechelska-Noworyta, M. Owinska and M. Hasik, J. Organomet. Chem., 2019, 898, 10 CrossRef.
- D. D. Roberts and M. G. McLaughlin, Org. Lett., 2021, 23, 4463–4467 CrossRef CAS PubMed.
- K.-H. Huang and M. Isobe, Eur. J. Org. Chem., 2014, 4733–4740 CrossRef CAS.
- S. Ding, L.-J. Song, Y. Wang, X. Zhang, L. W. Chung, Y.-D. Wu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 5632–5635 CrossRef CAS PubMed.
- J. B. Perales and D. L. Van Vranken, J. Org. Chem., 2001, 66, 7270–7274 CrossRef CAS PubMed.
- W. Gao, H. Ding, T. Yu, Z. Wang and S. Ding, Org. Biomol. Chem., 2021, 19, 6216–6220 RSC.
- X. Xie, X. Zhang, W. Gao, C. Meng, X. Wang and S. Ding, Commun. Chem., 2019, 2, 101 CrossRef.
- Y. Na and S. Chang, Org. Lett., 2000, 2, 1887–1889 CrossRef CAS PubMed.
- H. Yi, W. Mao and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 3575–3578 CrossRef CAS PubMed.
- O. Kitagawa, T. Suzuki and T. Taguchi, Tetrahedron Lett., 1997, 38, 8371–8374 CrossRef CAS.
- M. Rofoo, M.-C. Roux and G. Rousseau, Tetrahedron Lett., 2001, 42, 2481–2484 CrossRef CAS.
- J. J. Rojas, R. A. Croft, A. J. Sterling, E. L. Briggs, D. Antermite, D. C. Schmitt, L. Blagojevic, P. Haycock, A. J. P. White, F. Duarte, C. Choi, J. J. Mousseau and J. A. Bull, Nat. Chem., 2022, 14, 160–169 CrossRef CAS PubMed.
- J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150–12233 CrossRef CAS PubMed.
- S. C. Wilkinson, O. Lozano, M. Schuler, M. C. Pacheco, R. Salmon and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 7083–7086 CrossRef CAS PubMed.
- M. G. McLaughlin and M. J. Cook, Chem. Commun., 2011, 47, 11104–11106 RSC.
- D. Roberts and M. McLaughlin, Adv. Synth. Catal., 2022 DOI:10.1002/adsc.202200237.
- X. Zeng, C. Miao, S. Wang, C. Xia and W. Sun, Synthesis, 2013, 2391–2396 CAS.
- M. G. McLaughlin, C. A. McAdam and M. J. Cook, Org. Lett., 2015, 17, 10–13 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc03339a |
| This journal is © The Royal Society of Chemistry 2022 |