
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Utilizing a needle as a source of iron in synergistic dual photoredox catalytic generation of alkoxy radicals†
Alica
Ondrejková
 ,
Rickard
Lindroth
,
Göran
Hilmersson
and
Carl-Johan
Wallentin
*
,
Rickard
Lindroth
,
Göran
Hilmersson
and
Carl-Johan
Wallentin
*
Department of Chemistry and Molecular Biology, University of Gothenburg, 412 58 Gothenburg, Sweden. E-mail: carl.wallentin@chem.gu.se
First published on 4th August 2022
Abstract
A visible-light mediated alkoxy radical generation is described, which allows for a structurally divergent oxidative C(sp3)–H bond functionalization. This protocol employs a photoredox/iron dual catalysis allowing for an unprecedented chemoselective single-step transformation of alcohol derivatives giving access to two valuable sets of products, tetrahydrofurans and γ-bromoketones, under one set of conditions. Addition of iron, by slow corrosion of a needle, provides superior reaction efficiency as compared to various iron(III) complexes.
Oxygen-centered radicals are open-shell, highly reactive species that can participate in a variety of valuable reactions.1 Despite their rich synthetic possibilities, there is a limited number of reliable strategies for their generation and utilization in the construction of molecular complexity. Typically, facile generation of alkoxy radicals often requires stoichiometric quantities of very strong oxidants.2–7 Consequently, the most common approach was the employment of alkoxy radical precursors. However, the preparation and/or utilization of historically common alkoxy radical precursors often involves explosive reagents or strong oxidants,8,9 tedious synthesis10 or unstable nature of precursor itself,11 necessity of organotin initiator,10 UV irradiation or elevated temperatures12–14 which naturally diminished their synthetic potential. More recently, photocatalysis15–17 has emerged as a powerful platform for alkoxy radical generation under mild conditions. Recent work from Knowles group (Scheme 1B)18 has shown that hydroxyl group can be directly oxidized via reductive quenching of a photocatalyst, thereby generating an oxygen-centered radical. In efforts toward alkoxy radicals, research groups of Zhu (Scheme 1B)19,20 and Almqvist21 have developed protocols for efficient alkoxy radical initiated ring-opening of tertiary cycloalkanols via β-scission of cyclic C–C bonds. The research group of Chen,22 as well as our own laboratory,23 have recently reported an approach relying on oxidative quenching of the photocatalyst, utilizing hypervalent iodine(III) alkoxy radical precursors in 1,5-hydrogen atom transfer (1,5-HAT) mediated transformations (Scheme 1A).
 | ||
| Scheme 1 (A) Our previously reported etherification method utilizing hypervalent iodine(III) alkoxy radical precursors, (B) previously reported methods towards halogenated ketones utilizing alkoxy radicals, (C) visible-light mediated etherification and bromoketone synthesis utilizing alkoxy radicals and 1,5-HAT. | ||
As a part of our ongoing engagement in oxygen-centered radicals and photoredox catalysis, we have investigated the possibility to access these moieties utilizing N-alkoxythiazolethione precursors. Given their capacity as an efficient alkoxy radical source,24 we hypothesized that redox-active thiazolethione (Thi) functionality could be strategically employed in oxidative quenching of the photocatalyst, subsequently activating δ-C(sp3)–H position in new C–O bond-forming events. Herein, we present a new method that allows for a structurally divergent strategy providing the opportunity to selectively transform linear alcohols to cyclic ethers and linear γ-bromoketones by employing a synergistic dual catalytic system using a photocatalyst in combination with a needle acting as a source of Fe (Scheme 1C).
Initial efforts were focused on establishing an efficient 1,5-HAT system. For this, N-alkoxythiazolethione 1, derived from 4-phenylbuthanol, was chosen as our model substrate to allow for an efficient δ-C(sp3)–H abstraction together with subsequent oxidation and cyclization giving the corresponding tetrahydrofuran derivative. Bromotrichloromethane (CBrCl3) was chosen as external oxidant since it is known to oxidatively quench several common photoexcited catalysts.25 Extensive screening of photocatalysts, solvents, additives, and stoichiometry of the reagents was conducted (Table 1 and ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ru(bpy)3 (mol%) | CBrCl3 (equiv.) | Additive | Yielde (%) | |
| 2 | 3 | ||||
| a Reaction time 15 h, reaction temperature 25 °C. b Reaction time 6 h, reaction temperature 55 °C. c Reaction performed with a metallic needle inserted into the system. d Reaction performed in the dark. e Yields were determined by 1H NMR using ethylene carbonate as an internal standard. | |||||
| 1a | 5 | 1.5 | — | 62 | 43 |
| 2a | 5 | 10 | — | 23 | 64 |
| 3a | 5 | 20 | — | — | 69 |
| 4bc | 5 | 10 | — | — | 66 |
| 5bc | 5 | 10 | LiBr | 49 | 15 |
| 6bc | 5 | 10 | NBS | 28 | 9 |
| 7 | 5 | 10 | TBAB | — | 95 |
| 8bc | 5 | 1.5 | TBAB | 54 | 24 |
| 9 | 5 | 10 | TBAB | 72 | 19 |
| 10bd | 5 | 10 | TBAB | — | — |
| 11b | 0 | 10 | TBAB | — | — |

Surprisingly, initial experiments yielded a mixture of tetrahydrofuran 2 and bromoketone 3. This rather intriguing result opens for the possibility of a chemodivergent method providing access to two distinctively different structural features by simple tweaking of the conditions. Monitoring the reaction over time revealed that tetrahydrofuran 2 serves as an intermediate for bromoketone 3 formation (see ESI†). Consequently, we set out to optimize the conditions for both 2 and 3. The optimal loading of CBrCl3 towards tetrahydrofuran 2 was found to be 1.5 equiv. (62%, entry 1). If the loading was lowered, even after prolonged reaction time, the reaction did not proceed with full conversion. With 10 equiv. of CBrCl3 we were able to obtain satisfactory results for bromoketone 3 (64%, entry 2). Increasing the temperature to 55 °C increased the yield slightly, but more importantly, did so with a significantly shortened reaction time from 15 h to 6 h (entry 4). To further facilitate the ring-opening of 2, we reasoned that an external source of bromide would increase the yield of desired ketone 3. Various external bromide sources were evaluated (entries 5–7) and we were pleased to find that the addition of tetrabutylammonium bromide (TBAB) led to almost a quantitative 95% yield of 3 (entry 7). Throughout the initial optimization, discrepancies in the results were observed for several conditions. After meticulous investigation of all variables, we observed that having a metallic needle, used as a part of the experimental setup to keep the system under a N2 atmosphere, present during the entire reaction time, reproducible yield of 3 was obtained (entry 7). Employing identical conditions with the needle removed provided 2 in 72% yield (entry 9). The notable difference in reaction outcome can be explained by that earth abundant transition metals present in the needle entered the reaction solution and mediated the conversion of 2 to 3. Observation of slow corrosion of the metallic needle, likely due to formation of HBr during the progression of the reaction, is consistent with this assumption. In our previous efforts, we have observed that ring-opening of cyclic ethers can be facilitated under similar conditions, in the presence of trace amount of transition metals,26 which further supports the hypothesis of metal catalysis. Therefore, the metal composition of the needle was evaluated by XRF, and major metals present in the needle were tested in our system (see ESI†). Although the evaluated metal complexes (Fe[III], Ni[II], Cr[III]) mediated the conversion of 2 to 3, none could match the efficiency of introducing the needle into the system. ICP-MS analysis of the crude reaction mixture clearly showed that the main metals present in the needle indeed did corrode into the reaction mixture (see ESI†). Lastly, no products were detected in control experiments where the photocatalyst or blue light irradiation were excluded from the system supporting their necessity in the given transformation (entry 11 and 12).
With optimized conditions in hand, we explored the scope of N-alkoxythiazolethione derivatives. Initially, we dedicated our efforts toward primary N-alkoxythiazolethione derivatives (Scheme 2). Aromatic substituents with various electronic properties are well tolerated for both transformations. Both electron-donating and electron-withdrawing aryl substituents were evaluated, and we were pleased to obtain both tetrahydrofurans 2a–2j and corresponding bromoketones 3a–3j derived from primary alcohols in good to excellent yields. Worth noting is that a p-nitro functionality, which is very often found to be problematic in photoredox catalysis, was compatible with our conditions and gave rise to tetrahydrofuran 2c in 54% yield and the corresponding bromoketone 3c in high 78% yield. Aromatics bearing p-halo substituents gave rise to tetrahydrofurans 2f, 2h and 2j in high yields (94%, 84% and 79%) and 2g with p-chloro substituted phenyl ring in slightly lower 55% yield. In terms of their subsequent conversion to the corresponding bromoketones 3f–3i, these were obtained in good to high yields (65–86%). Additionally, the electron rich heteroaromatic thiophene moiety was also compatible with this transformation and delivered tetrahydrofuran 2j and the corresponding ketone 3j in good 58% and 50% yields, respectively. These examples illustrate that the process has a broad scope in terms of the electronic nature of the arene moiety.
 | ||
Scheme 2 Scope of primary N-alkoxythiazolethione derivatives. Reaction conditions: 1 (0.1 mmol), Ru(bpy)3(PF6)2 (5 mol%), CBrCl3 (10 equiv.), TBAB (1.1 equiv.) in CH3CN (0.05 M), blue LED irradiation for 6 h at 55 °C, a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) needle inserted into the vial, breaction conducted on 1 mmol scale. Yields refer to isolated products. needle inserted into the vial, breaction conducted on 1 mmol scale. Yields refer to isolated products. | ||
Next, we turned to explore the scope of secondary alkoxy radicals where the 4-phenylbuthanol fragment of the molecule was retained while the α-substituent of the alcohol was alternated (Scheme 3). We were pleased to find that all investigated secondary N-alkoxythiazolethiones led to desired tetrahydrofurans 2k–2r as well as corresponding bromoketones 3k–3r in good to high yields. Both methyl and sterically demanding isobutyl substituted substrates provided tetrahydrofurans 2k and 2l in 68% and 70% yields, respectively. Their corresponding bromoketones 3k and 3l were obtained with yields of 64% and 65%, respectively. For longer butyl substituents, two possible pathways for 1,5-HAT are available. However, a benzylic C(sp3)–H bond is more activated towards 1,5-HAT, resulting in more thermodynamically favoured carbon-centered radical. Indeed, tetrahydrofuran 2m was isolated as a major regioisomer in 58%. The formation of the other possible regioisomer was observed in negligible quantity. Bromoketone 3m was obtained in slightly lower 48% yield. Considerably bulkier phenethyl N-alkoxythiazolethione yielded desired tetrahydrofuran 2n in 75% and bromoketone 3n in 65% yields. Next, we moved our attention towards α-aryl-substituted N-alkoxythiazolethiones (1o–1r). The transformation to tetrahydrofurans 2o, 2p and 2r proceeded smoothly in 69%, 59% and 63% yields, respectively. The reaction of α-phenyl substituted N-alkoxythiazolethione alcohol and its p-chloro derivative, proceeded towards bromoketone 3p and 3r in 70% and 62% yields, respectively, with reaction time shortened to 3 h. Longer reaction time converted bromoketones 3p and 3r into their corresponding dehalogenated derivatives. This result can be rationalized by the ability of Ru(II) to initiate visible-light mediated dehalogenation in the presence of HCCl3 as H-atom donor.15,27 Electron-rich o-methoxy substituted derivative provided trichloromethyl functionalized tetrahydrobenzoxepine 3o in 54% yield (see ESI†).
 | ||
| Scheme 3 Scope of secondary N-alkoxythiazolethione derivatives. Reaction conditions: 1 (0.1 mmol), Ru(bpy)3(PF6)2 (5 mol%), CBrCl3 (10 equiv.), TBAB (1.1 equiv.) in CH3CN (0.05 M), blue LED irradiation for 6 h at 55 °C, aneedle inserted into the vial, breaction time 3 h, cregioisomeric ratio. Yields refer to isolated products. | ||
To demonstrate the synthetic utility of γ-bromoketones, we converted 4-bromo-1-phenylbutane-1-one 3 to various structural entities of general significance (Scheme 4). Asymmetric reduction of 3, followed by ring closure generated (S)-2-phenyltetrahydrofuran 4 in 87% yield with 86% ee. Functionalized 2-methyl-2-phenyltetrahydrofuran 5 was obtained in 85% yield whereas cyclopropane 6 and dihydrooxazine 7 were generated in nearly quantitative yields.
 | ||
| Scheme 4 Demonstration of synthetic applications of γ-bromoketones. | ||
A plausible mechanism for the developed process is depicted in Scheme 5. The photoexcited catalyst RuII*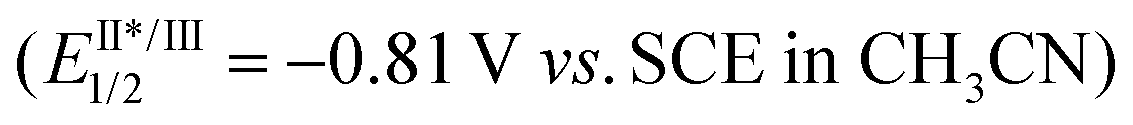 15 is oxidatively quenched by CBrCl3 to afford RuIII along with ˙CCl3 and Br−. Addition of ˙CCl3 radical to the thione sulfur, followed by the homolytic cleavage of the O–N bond liberates a desired oxygen-centered radical. Subsequently, the alkoxy radical undergoes 1,5-HAT generating a stabilized benzylic carbon-centered radical. This radical intermediate (Eox = ca. 0.35 V vs. SCE in CH3CN)28 can be further oxidized by RuIII (EIII/II1/2 = 1.29 V vs. SCE in CH3CN)15 into its corresponding carbocation. This oxidation event returns the photocatalyst to its ground state. The new C–O bond is formed via intramolecular cyclization yielding the tetrahydrofuran derivative. Further transformation into the corresponding bromoketone proceeds via hydrogen abstraction mediated by another molecule of ˙CCl3 radical affording cyclic radical intermediate (Eox = ca. 0.16 V vs. SCE in CH3CN)28 which can be oxidized by RuIII (or alternatively CBrCl3) into the corresponding cyclic carbocation. This process is further facilitated by an iron catalyzed generation of both ˙CCl3 and carbocation under blue-light irradiation.26 Lastly, the ring-opening event of carbocation by bromide ion provides the final bromoketone.
15 is oxidatively quenched by CBrCl3 to afford RuIII along with ˙CCl3 and Br−. Addition of ˙CCl3 radical to the thione sulfur, followed by the homolytic cleavage of the O–N bond liberates a desired oxygen-centered radical. Subsequently, the alkoxy radical undergoes 1,5-HAT generating a stabilized benzylic carbon-centered radical. This radical intermediate (Eox = ca. 0.35 V vs. SCE in CH3CN)28 can be further oxidized by RuIII (EIII/II1/2 = 1.29 V vs. SCE in CH3CN)15 into its corresponding carbocation. This oxidation event returns the photocatalyst to its ground state. The new C–O bond is formed via intramolecular cyclization yielding the tetrahydrofuran derivative. Further transformation into the corresponding bromoketone proceeds via hydrogen abstraction mediated by another molecule of ˙CCl3 radical affording cyclic radical intermediate (Eox = ca. 0.16 V vs. SCE in CH3CN)28 which can be oxidized by RuIII (or alternatively CBrCl3) into the corresponding cyclic carbocation. This process is further facilitated by an iron catalyzed generation of both ˙CCl3 and carbocation under blue-light irradiation.26 Lastly, the ring-opening event of carbocation by bromide ion provides the final bromoketone.
 | ||
| Scheme 5 Proposed mechanism based on generated data. | ||
In summary, this study set out to develop a new method for alkoxy radicals generation under mild, photon-driven conditions, for the purpose of selective C(sp3)–H functionalization. By modulating the presence of an earth-abundant metal catalyst in combination with a common photocatalyst, we have shown that it is possible to selectively access cyclic ethers and γ-bromoketone derivatives under visible-light irradiation conditions. The protocol is operationally simple and allows for synthetic diversity for simple alcohols under one set of conditions, giving rapid access to a broad collection of structurally diverse compounds.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed.
- C. C. González, A. R. Kennedy, E. I. León, C. Riesco-Fagundo and E. Suárez, Angew. Chem., Int. Ed., 2001, 40, 2326–2328 CrossRef.
- C. G. Francisco, C. González Martín and E. Suárez, J. Org. Chem., 1998, 63, 2099–2109 CrossRef CAS.
- H. Suginome and J. B. Wang, J. Chem. Soc., Chem. Commun., 1990, 22, 1629–1631 RSC.
- L. Brewster Young and W. S. Trahanovsky, J. Am. Chem. Soc., 1969, 91, 5060–5068 CrossRef.
- A. Clerici, F. Minisci, K. Ogawa and J.-M. Surzur, Tetrahedron Lett., 1978, 19, 1149–1152 CrossRef.
- K. Heusler, Tetrahedron Lett., 1964, 5, 3975–3982 CrossRef.
- J. E. Leffler, Chem. Rev., 1949, 45, 385–417 CrossRef CAS.
- C. H. Robinson, O. Gnoj, A. Mitchell, R. Wayne, E. Townley, P. Kabasakalian, E. P. Oliveto and D. H. R. Barton, J. Am. Chem. Soc., 1961, 83, 1771–1772 CrossRef CAS.
- S. Kim, T. A. Lee and Y. Song, Synlett, 1998, 471–472 CrossRef CAS.
- A. L. J. Beckwith and B. P. Hay, J. Am. Chem. Soc., 1988, 110, 4415–4416 CrossRef CAS.
- V. Malatesta and J. C. Scaiano, J. Org. Chem., 1982, 47, 1455–1459 CrossRef CAS.
- Ch. Walling and A. Padwa, J. Am. Chem. Soc., 1963, 85, 1593–1597 CrossRef CAS.
- D. J. Pasto and F. Cottard, Tetrahedron Lett., 1994, 35, 4303–4306 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794–10797 CrossRef CAS PubMed.
- D. Wang, J. Mao and C. Zhu, Chem. Sci., 2018, 9, 5805–5809 RSC.
- X. Wu and C. Zhu, Chem. Rec., 2018, 18, 587–598 CrossRef CAS PubMed.
- D. Petzold, P. Singh, F. Almqvist and B. König, Angew. Chem., Int. Ed., 2019, 58, 8577–8580 CrossRef CAS PubMed.
- K. Jia, F. Zhang, H. Huang and Y. Chen, J. Am. Chem. Soc., 2016, 138, 1514–1517 CrossRef CAS PubMed.
- A. Rivero, P. Fodran, A. Ondrejková and C.-J. Wallentin, Org. Lett., 2020, 22, 8436–8440 CrossRef CAS PubMed.
- J. Hartung, Eur. J. Org. Chem., 2001, 619–632 CrossRef CAS.
- J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed.
- R. Lindroth, A. Ondrejková and C.-J. Wallentin, Org. Lett., 2022, 24, 1662–1667 CrossRef CAS PubMed.
- J. Ho, J. Zheng, R. Meana-Pañeda, D. G. Truhlar, E. Jung Ko, G. Paul Savage, C. M. Williams, M. L. Coote and J. Tsanaktsidis, J. Org. Chem., 2013, 78, 6677–6687 CrossRef CAS PubMed.
- D. D. M. Wayner, D. J. McPhee and D. Griller, J. Am. Chem. Soc., 1988, 110, 132–137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc03262g |
| This journal is © The Royal Society of Chemistry 2022 |