
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeriodate anions as a halogen bond donor: formation of anion⋯anion dimers and other adducts†‡
Miriam
Calabrese
 a,
Andrea
Pizzi
*a,
Andrea
Daolio
a,
Antonio
Frontera
*b and
Giuseppe
Resnati
*a
a,
Andrea
Pizzi
*a,
Andrea
Daolio
a,
Antonio
Frontera
*b and
Giuseppe
Resnati
*a
aNFMLab, Department Chemistry, Materials, and Chemical Engineering “Giulio Natta”, Politecnico di Milano, via L. Mancinelli 7, I-20131 Milano, Italy. E-mail: andrea.pizzi@polimi.it; giuseppe.resnati@polimi.it
bDepartment Chemistry, Universitat de les Illes Balears, Crta, de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain. E-mail: toni.frontera@uib.es
First published on 21st July 2022
Abstract
Single crystal X-ray analyses show that iodine in pyridinium periodates acts as a halogen bond (HaB) donor forming short and almost linear contacts with neutral and anionic electron donors. A combination of QTAIM and NCIplot computational tools proves the attractive nature of these contacts.
The recognition of the anisotropic distribution of the electron density around bonded atoms1 allows for a reliable prediction of the occurrence of a variety of noncovalent interactions and paved the way to a systematic rationalization of many of them.2
Many noncovalent forces related to the electrophilic behaviour of covalently bonded atoms have been identified and categorized3 within the frame of σ-holes,4i.e. the regions of depleted electron density opposite to single covalent bonds. Elements of the p block of the periodic table have been investigated after this viewpoint, and according to the IUPAC definition, σ-hole interactions given by elements of groups 17 and 16 can be named halogen bonds (HaBs)5 and chalcogen bonds (ChBs),6 respectively. Recently, an analogous mindset was developed also for interactions formed by elements of the d block, specifically elements of groups 7 (matere bond, MaB),7 8 (osme bond, OmB),8 11 (coinage bond, CiB),9,10 and 12 (spodium bond, SpB).11
For group 8 elements, Os⋯N/O short contacts have been identified experimentally in neutral adducts between OsO4 (electrophile) and pyridine/pyridine N-oxide derivatives (nucleophile). These contacts are on the elongation of O–Os covalent bonds, as typical for σ-hole interaction.8 Results of theoretical investigations, the slight distortion of the OsO4 tetrahedral geometry on adduct formation, and the high oxidation state of osmium, consistently led to the inference that the short contacts involving the metal are not of coordinative nature. A similar behaviour has been observed also in tetrahedral derivatives of group 7 elements as perrhenate and permanganate anions can behave as electrophiles.7
Many reports describe the reliability of tetrahedral and neutral σ-hole donors wherein the donor atom is a p block element, e.g., a carbon,12 germanium,13 and tin14 atom. We decided to make a major step forward and to identify tetrahedral and anionic σ-hole donors wherein the donor atom is a p block element. Moving from the interest in hypervalent iodine compounds,15–17 we decided to explore the σ-hole donor ability of anionic iodine in its highest oxidation state, i.e. in the periodate anion. This anion was exploited as a tridentate nucleophile in the construction of (6,3) halogen-bonded networks on self-assembly with 1,4-diiodotetrafluorobenzene acting as a bidentate electrophile.18 However, to the best of our knowledge, the potential of the electrophilic behaviour of iodine in the periodate anion has never been investigated in recognition phenomena. Here, we report that in the solid the iodine atom of pyridinium periodates 1–4 (Scheme 1) forms short contacts19 with neutral and negatively charged electron rich atoms (Fig. 1). Theoretical calculations prove the attractive nature of these contacts and allow for periodate anions to be added to the palette of hypervalent halogens functioning as HaB donors. Pyridinium ring substituents in salts 1–4 are in meta and para positions and have different steric and geometric features. The HaB occurrence independent of these differences suggests that periodate anions may function as robust and reliable HaB donor tectons.
 | ||
| Scheme 1 Chemical structure of the pyridinium periodates 1–4. | ||
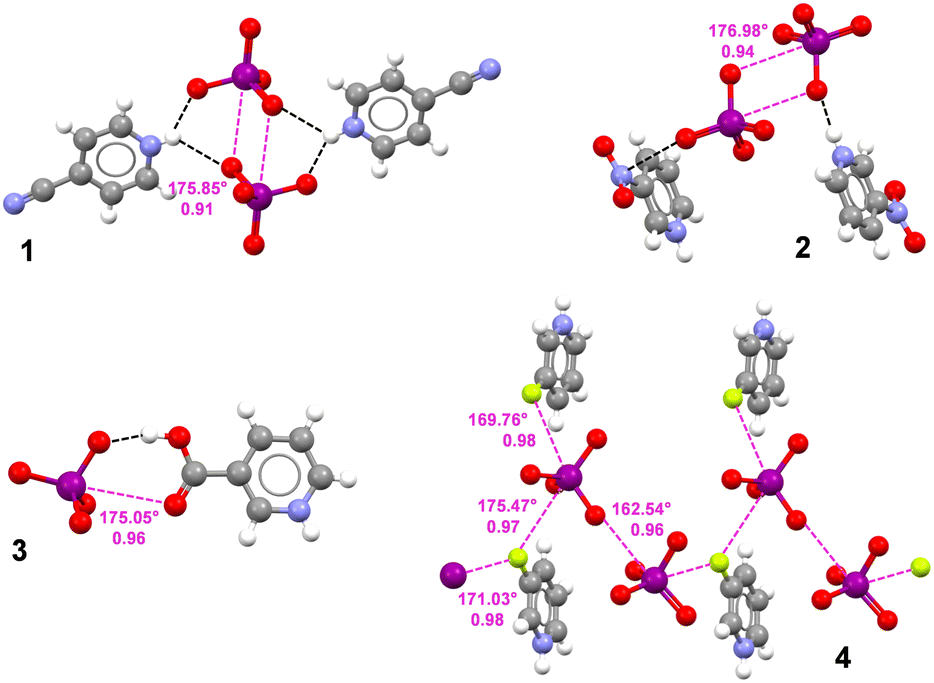 | ||
| Fig. 1 Ball and stick representation (Mercury, 2022.2.0) of crystal structures 1-4 evidencing the HaBs involving the periodate anion (purple dotted lines). Nc values of HaBs and O–I⋯O/F angles are given close to the HaBs. Selected HBs (black dotted lines) are reported for salts 1–3. Colour code: whitish, hydrogen; grey, carbon; red, oxygen; indigo, nitrogen; purple, iodine; yellow, fluorine. | ||
Single crystal X-ray analyses of 4-cyanopyridinium periodate (1) and 3-nitropyridinium periodate (2) (Fig. 1) display anion⋯anion dimers where each IO4− unit functions as both a HaB donor and acceptor and two IO4− units self-assemble via an antiparallel pairing. This recognition motif is commonly shown by σ- and π-hole based tectons wherein the donor and acceptor atoms are directly bonded to each other.7,9,10 Consistent with the typical HaB features, I⋯O short contacts occur on the extension of a O–I covalent bond, namely the O–I⋯O angles are very close to linearity (175.85° in 1 and 176.98° in 2). The observed I⋯O separations are far below the sum of van der Waals radii20 of the involved atoms, with normalized contacts (Nc)21 which are notably small for anion⋯anion interactions (0.91 in 1 and 0.94 in 2). In both structures, hydrogen bonds (HBs) between the cationic unit and the IO4− oxygen atoms are present and exert an important role in withdrawing negative charge from the anion, as confirmed by computations. Of note, in 2 a strong π-hole interaction involving the nitrogen atom of the nitro group and one oxygen atom of the anion further contributes to the IO4− negative charge dissipation.
In order to investigate the cation effect on the electrophilicity of iodine in IO4−, we performed the analysis of the molecular electrostatic potential (MEP) of a naked periodate anion and of compound 1, used as exemplifying pyridinium salt (Fig. 2). For the naked anion, the MEP values are negative on the entire surface, as expected. However, it can be observed that the distribution of the electron density is anisotropic, disclosing the existence of four symmetric (negative) σ-holes on the extensions of the O–I bonds. Remarkably, in compound 1 where the cation is H-bonded to one periodate O-atom, the MEP value at the iodine's σ hole changes drastically becoming positive (Fig. 2b). Obviously, the MEP maximum is located in the cationic part of the salt (aromatic H-atoms, +55.0 kcal mol−1) and the minimum at the O atoms of the anion (−37.0 kcal mol−1).
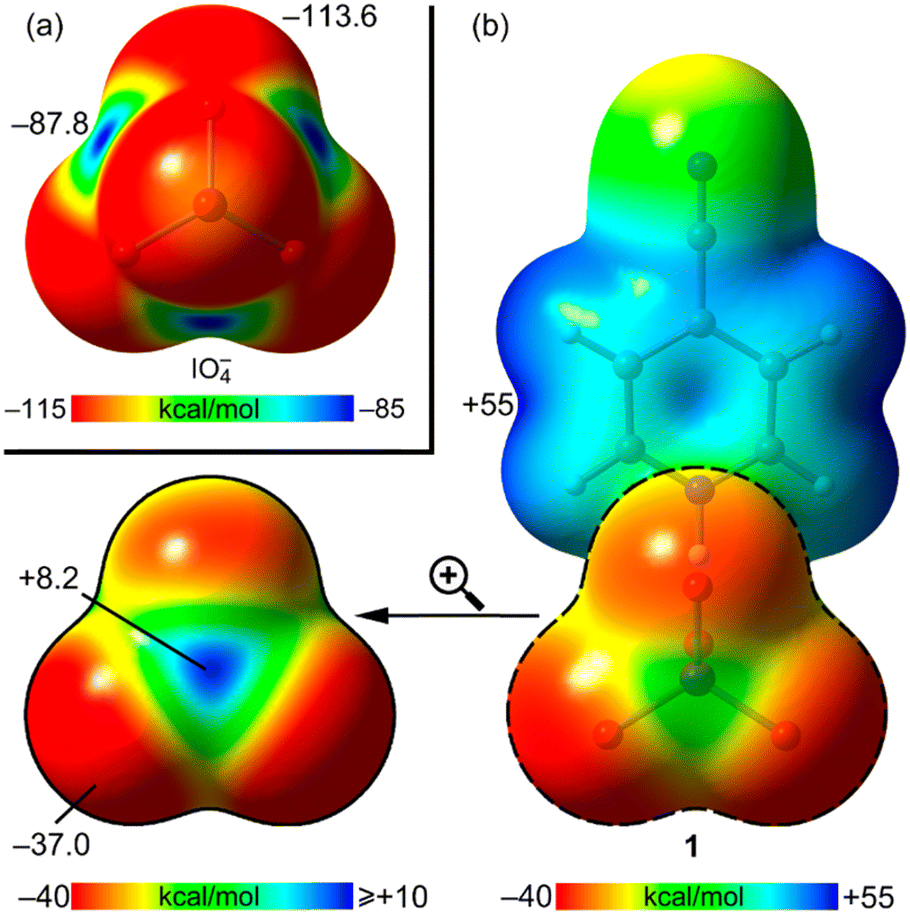 | ||
| Fig. 2 MEP surfaces (isosurface 0.001 a.u.) of the periodate anion (a) and compound 1 (b) optimized at the PBE0-D3/def2-TZVP level of theory. Values at some points of the surfaces are indicated in kcal mol−1. | ||
This analysis anticipates the attractive nature of the I⋯O contacts reported in Fig. 1. Moreover, the HB driven withdrawal of negative charge from the anion is expected to be more important in the X-ray structure, where the anion participates in more than one HB (or π-hole) interaction.
In 3-carboxypyridinium periodate (3) and 3-fluoropyridinium periodate (4), the IO4− moiety acts as an electron density acceptor, i.e. HaB donor, towards neutral nucleophilic atoms. In detail, in 3 the carbonyl oxygen of the carboxylic acid gets close to iodine (I⋯O separation 349 pm, Nc 0.96, O–I⋯O angle 175.05°). This almost ideal geometry of the HaB is favoured by a strong HB between an IO4− oxygen and the hydroxyl group of the carboxylic functionality. In 3-fluoropyridinium periodate 4, an I⋯O HaB assembles two periodate anions into a supramolecular dimer (I⋯O distance 351.6 pm, Nc 0.96; O–I⋯O angle 162.54°). In this dimer the iodine atoms of the periodate units acting as HaB acceptor and donor also function as bidentate and monodentate HaB donors, respectively. They accept electron density from two crystallographically different fluorine atoms and the I⋯F separations are 349.2 pm (Nc 0.97) and 351.8 pm (Nc 0.98) on the bidentate iodine (respective O–I⋯F angles are 175.47° and 169.76°) and 353.2 pm (Nc 0.98) on the monodentate iodine (respective O–I⋯F angle is 171.03°).
Further theoretical studies have been carried out in order to assess the attractive nature of the short contacts involving the periodate anions and in order to confirm the ability of iodine in these anions to act as an electrophile, i.e., HaB donor. First, we used two methods based on the topology of the electron density (see ESI‡ for details), namely the quantum theory of atoms in molecules (QTAIM) and the noncovalent interaction plot (NCIPlot). Combined information from the two methods is useful to characterize noncovalent interactions in real space. In particular, the reduced density gradient (RDG) isosurfaces not only provide information about which intermolecular regions interact but also if the interaction is attractive or repulsive. Blue-green colours, meaning (signλ2)ρ < 0, are used for attractive and red-yellow colours for repulsive interactions [(signλ2)ρ > 0]. The results are given in Fig. 3 for compounds 1 and 4 (see ESI,‡ Fig. S6, for similar assemblies of compounds 2 and 3). In 1, we have analysed the self-assembled dimer already commented above and shown in Fig. 1. Apart from the HB network, the QTAIM analysis confirms the existence of the reciprocal I⋯O HaBs that are characterized by bond critical points (CPs, small spheres) and bond paths interconnecting the I and O atoms. Moreover, the interactions are characterized by bluish RDG isosurfaces, thus evidencing their attractive nature. The QTAIM analysis also shows the existence of a O⋯O contact characterized by a bond CP and bond path interconnecting both O-atoms. The colour of the NCIplot RGD isosurface is yellow around the bond CP, thus suggesting that this interaction is repulsive and most likely compensated by other interactions. In fact, the dimerization energy is very large (it has been computed for the coupling of two 4-cyanopyridinium periodate ion pairs), ΔE1 = −38.9 kcal mol−1, due to the strong nature of the charge assisted HBs, as confirmed by the blue colour of the RGD isosurface of the NH⋯O HBs and the HaBs. Similar interaction energies have been found for compounds 2 and 3 (see Fig. S6, ESI‡).
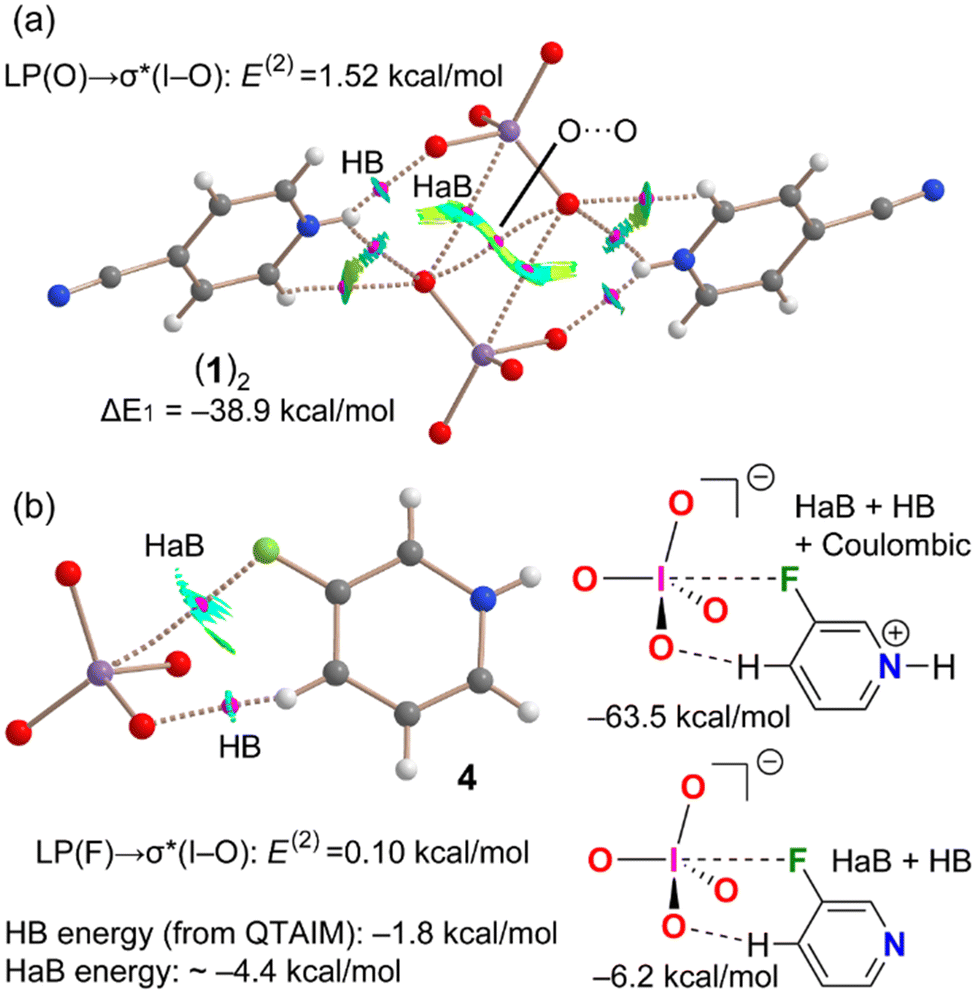 | ||
| Fig. 3 (a) Combined QTAIM/NCIPlot for a hetero tetramer of compound 1. Only intermolecular interactions are represented by bond CPs (in fuchsia) and bond paths (dashed lines). The interaction energy was evaluated as a dimer of two ion pairs. (b) Combined QTAIM/NCIPlot of compound 4 (left). Models used to evaluate the contribution of the HaB (right). The energy of the HB was evaluated using the equation E = 1/2 × Vr. | ||
Combined QTAIM/NCIPlot analysis has been also performed for 3-fluoropyridinium periodate salt (4) (Fig. 3b). Interestingly, it evidences the existence of two anion-cation contacts. One is an HB between an aromatic hydrogen and a periodate oxygen and is characterized by the corresponding bond CP, bond path, and blue isosurface (see Fig. S5, ESI‡). The other one is the HaB between the F atom and the I atom, also characterized by a bond CP, bond path, and a blue RGD isosurface. The binding energy is very large due to the pure Coulombic attraction between the counterions (ΔE2 = −63.5 kcal mol−1). In an effort to estimate the HaB energy free from the electrostatic effects resulting from ion pairing, we have used a model employing 3-fluoropyridine (Fig. 3, bottom-right).22 The interaction is drastically reduced to ΔE3 = −6.2 kcal mol−1, which corresponds to the contribution of both halogen and hydrogen bonds. The energy of the HB has been estimated using the potential energy density at the bond CP and the equation proposed by Espinosa et al.24 The computed energy of the C–H⋯O HB is −1.8 kcal mol−1 and that of the O–I⋯F HaB is −4.4 kcal mol−1, in line with conventional HaB energies reported in the literature.23
For compound 3, we have compared the geometric and energetic features of the optimized geometry with the experimental one (Fig. 4). It can be observed that the two geometries are in acceptable agreement, taking into consideration that the DFT calculation is in the gas phase and uses an isolated dimer. In the gas phase there is an elongation of the carboxylate OH bond. Interestingly, the dimerization energies are similar using the optimized or experimental geometries, thus suggesting that the HaBs observed in the solid state are not due to packing effects. The combined QTAIM/NCIPlot analysis of optimized compound 3 is represented in Fig. 4c. The bond CP, bond path, and blue RDG isosurface characterizing the O–I⋯O HaB confirm its attractive nature.
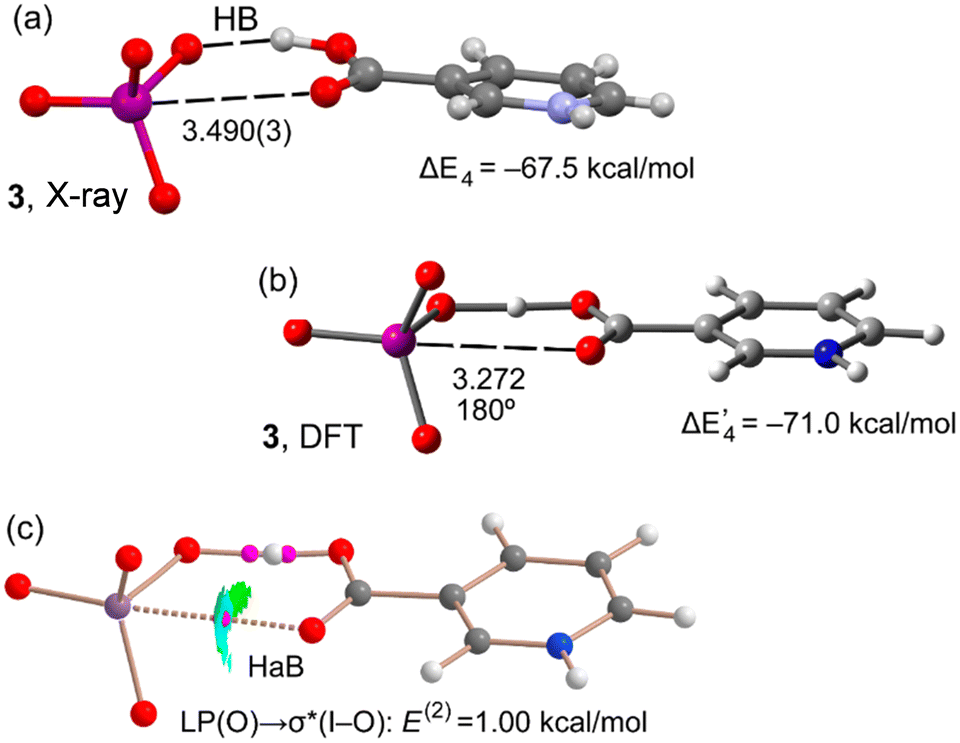 | ||
| Fig. 4 (a) X-ray geometry of compound 3 and its cation⋯anion interaction energy. Distances in Å. (b) DFT optimized geometry of 3 and its interaction energy. (c) Combined QTAIM and NCIplot of the optimized compound 3. Distances in Å. | ||
The natural bond orbital (NBO) analysis has been used in the assemblies of compounds 1, 3 and 4 to investigate if the LP → σ* orbital contribution common in σ-hole interactions exists in the HaBs involving a periodate anion. Interestingly, in all cases we have found either LP(O) → σ*(I–O) or LP(F) → σ*(I–O) orbital donor–acceptor contributions (see Fig. 3 and 4) which range from very small in compound 4 (0.1 kcal mol−1) to modest in 1 (1.52 kcal mol−1). Such small contributions are likely due to the anionic nature of the HaB donor and suggest that the HaB described herein are dominated by electrostatic effects.
Finally, we have also optimized the periodate dimer [IO4⋯IO4]2− taking into consideration solvent effects (in the gas phase the anions separate to infinitum) and using two possible orientations (Fig. 5), as observed in crystalline compounds 1–4 (see Fig. 1 and ESI‡). It is worthy to comment that the centrosymmetric dimer (observed in compounds 1, 2, Fig. 1 and Fig. S6, ESI‡) exhibiting two I⋯O HaBs is more favourable than the other one (observed in 4) where only one HaB contact is formed. Moreover, in the centrosymmetric dimer the HaB distance is shorter. It is worthy to highlight that the anion⋯anion HaB interaction is attractive in water, thus suggesting that the electrostatic repulsion of the anion⋯anion dimer can be counter-balanced by a convenient environment.
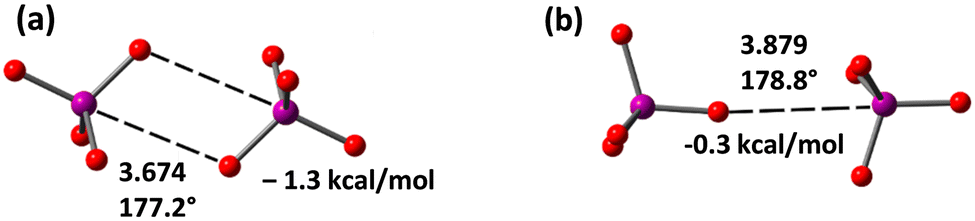 | ||
| Fig. 5 Optimized structures of two [IO4⋯IO4]2− dimers exhibiting two (a) or one (b) HaBs. Distances in Å. The dimerization energies in water are indicated. | ||
The Crystallographic Structural Database (CSD) contains only four compounds where the periodate anion functions as a HaB donor (see ESI‡ for greater details). These crystal structures seem to confirm theoretical indications that the involvement of periodate oxygen atoms in H⋯O HB formation plays a key role in unmasking the HaB donor potential of iodine by dissipating the negative charge of IO4− and making positive the electrostatic potential at iodine σ-holes.25,26 Interestingly, in imidazolium periodate, an improper molecular ferroelectric compound,27 IO4− anions are involved in a tight network of HBs and further self-assemble into anionic infinite chains very similar to those observed in perrhenate salts.7
In conclusion, the crystallographic analyses of some periodate salts show that iodine forms short and linear contacts with electron donor atoms. These interactions can be rationalized as HaBs and the ability of periodate iodine to act as an electrophile is robust enough to drive the formation of anion-anion adducts. Theoretical calculations reveal that in H-bonded periodate anions, the surface electrostatic potential at iodine σ-holes is positive, namely adequate for interacting with electron donors. A combination of QTAIM and NCIplot analyses proves the attractive nature of the short contacts observed in the studied crystals where periodate anions are the HaB donors. DFT calculations disclose that the HaB driven anion⋯anion interaction between periodate anions may exist also in solution.
A. F. acknowledges MICIU/AEI of Spain (PID2020-115637GB-I00, FEDER funds) for financial support and the CTI (UIB) for computational facilities. G.R. thanks PRIN-2020, project NICE, no. 2020Y2CZJ2.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- P. Politzer, K. E. Riley, F. A. Bulat and J. S. Murray, Comput. Theor. Chem., 2012, 998, 2–8 CrossRef CAS
.
-
S. Scheiner, Noncovalent Forces, Springer, 2015 Search PubMed
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Cryst. Growth Des., 2014, 14, 2697–2702 CrossRef CAS
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS
- C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef CAS
- A. Daolio, A. Pizzi, G. Terraneo, A. Frontera, G. Resnati, A. Daolio, A. Pizzi, G. Terraneo, G. Resnati and A. Frontera, Chem. Phys. Chem., 2021, 22, 2281–2285 CrossRef CAS PubMed
- A. Daolio, A. Pizzi, M. Calabrese, G. Terraneo, S. Bordignon, A. Frontera and G. Resnati, Angew. Chem., Int. Ed., 2021, 60, 20723–20727 CrossRef CAS PubMed
- A. Daolio, A. Pizzi, G. Terraneo, M. Ursini, A. Frontera and G. Resnati, Angew. Chem., Int. Ed., 2021, 60, 14385–14389 CrossRef CAS PubMed
- A. Pizzi, M. Calabrese, A. Daolio, M. Ursini, A. Frontera and G. Resnati, CrystEngComm, 2022, 24, 3846–3851 RSC
- A. Bauzá, I. Alkorta, J. Elguero, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2020, 59, 17482–17487 CrossRef PubMed
- A. Daolio, P. Scilabra, G. Terraneo and G. Resnati, Coord. Chem. Rev., 2020, 413, 213265 CrossRef CAS
- P. Scilabra, V. Kumar, M. Ursini and G. Resnati, J. Mol. Model., 2018, 24, 37 CrossRef PubMed
- A. Bauzá, S. K. Seth and A. Frontera, Coord. Chem. Rev., 2019, 384, 107–125 CrossRef
- F. Heinen, E. Engelage, C. J. Cramer and S. M. Huber, J. Am. Chem. Soc., 2020, 142, 8633–8640 CrossRef CAS PubMed
- V. V. Suslonov, N. S. Soldatova, P. S. Postnikov, G. Resnati, V. Yu Kukushkin, D. M. Ivanov and N. A. Bokach, Cryst. Growth Des., 2022, 22, 2749–2758 CrossRef CAS
- G. Cavallo, J. S. Murray, P. Politzer, T. Pilati, M. Ursini and G. Resnati, IUCrJ, 2017, 4, 411–419 CrossRef CAS PubMed
- A. Abate, J. Martí-Rujas, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Cryst. Growth Des., 2011, 11, 4220–4226 CrossRef CAS
- The term short contact, or close contact, is used to denote interactions shorter than the sum of van der Waals dadii of involved atoms (Batsanov's values were used, ref. 20).
- S. S. Batsanov, Inorg. Mater., 2001, 37, 1031–1046 Search PubMed
- The “Normalized Contact” Nc for an interaction involving atoms i and j is the ratio Dij/(RvdW,i + RvdW,j) where Dij is the experimental distance between i and j and RvdW,i and RvdW,j are the van der Waals radii of i and j.
- The use of fluoropyridine as a model for fluoropyridium in order to assess interaction energies is associated with approximations, e.g., the 4-H⋯O HB is weaker in the employed model system than in 4 (wherein the 4-H is more acidic), F is more prone to act as HaB acceptor in the model system than in 4 but iodine is less prone to act as HaB donor (as the anion negative charge is less effectively dissipated by the weaker HB). The usefulness of the employed model is nevertheless confirmed by the reliability of obtained evergies (ref. 23).
- V. Oliveira, E. Kraka and D. Cremer, Phys. Chem. Chem. Phys., 2016, 18, 33031–33046 RSC
- E. Espinosa, C. Lecomte and E. Molins, Chem. Phys. Lett., 1999, 300, 745–748 CrossRef CAS
- J. S. Murray and P. Politzer, Chem. Phys. Chem., 2021, 22, 1201–1207 CrossRef CAS PubMed
- J. S. Murray and P. Politzer, Crystals, 2020, 10, 76 CrossRef
- Y. Zhang, H. Y. Ye, H. L. Cai, D. W. Fu, Q. Ye, W. Zhang, Q. Zhou, J. Wang, G. L. Yuan and R. G. Xiong, Adv. Mater., 2014, 26, 4515–4520 CrossRef CAS PubMed
Footnotes |
| † Dedicated to the memory of Peter Politzer; the friend and the scientist. |
| ‡ Electronic supplementary information (ESI) available: Synthesis, spectroscopic, and crystallographic data of the examined adducts; details on computational analyses. CCDC 2168128–2168131. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc03191d |
| This journal is © The Royal Society of Chemistry 2022 |