
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalyzed decarboxylation of oxamic acids under near-infrared conditions†
Ikechukwu Martin
Ogbu
 ,
Dario M.
Bassani
,
Frédéric
Robert
and
Yannick
Landais
*
,
Dario M.
Bassani
,
Frédéric
Robert
and
Yannick
Landais
*
University of Bordeaux, Institute of Molecular Sciences (ISM), UMR-CNRS 5255, 351, Cours de la Libération, 33405 Talence Cedex, France. E-mail: yannick.landais@u-bordeaux.fr
First published on 8th July 2022
Abstract
Photocatalyzed oxidative decarboxylation of oxamic acids under near-infrared irradiation using Os(bptpy)2(PF6)2 as catalyst is reported. The reaction was applied to the synthesis of urethanes and heterocyclic amides. Mechanistic studies and comparative penetration depths between the NIR and the visible light mediated processes are discussed.
There is a growing interest in the use of near infrared (NIR) light in photocatalysis due to its promising advantages over UV-visible light (VL) counterparts.1 Although VL photocatalysis has shown its efficiency,2 its low penetration depth may hamper process scalability and productivity, adding to long reaction times inherent to photochemical processes. Recently, suitably-designed reactors covered with LEDs have been designed to improve the efficacy of VL-photoredox processes,3 but problems become more acute when viscous or heterogeneous mixtures are irradiated and a large amount of light is scattered by solid particles. In contrast to VL, red and NIR light possesses the ability to penetrate much deeper into a range of media (several cm), including polymers and biological tissues, which do not absorb or scatter at such wavelengths (600–1000 nm).4 On the negative side, NIR light can only provide low-energy excited states (∼35 kcal mol−1) which cannot promote electron or energy transfer processes required in many organic reactions.
Recent advances in this field have demonstrated that low-energy NIR can nonetheless be up-converted into blue light, and thus photoredox catalysis, through triplet–triplet annihilation up-conversion (TTA-UC),5 in which two low-energy photons are ultimately converted into a higher energy photon. Various sensitizer/annihilator pairs for TTA-UC of deep red/NIR light to blue light have been reported and some of them used for organic synthesis.6 Amongst the most efficient sensitizers, Pt and Pd catalysts have attracted some attention.5 Recently, Kimizuka and co-workers unveiled the use of Os(bptpy)2(PF6)2 complex (Fig. 1) for TTA-UC from NIR-to-blue light with a large anti-Stokes shift (0.97 eV) and quantum yield (ϕUC′) = 2.7%.7 More interestingly, this osmium(II) complex allows the direct population of triplet state from the ground state, a normally spin-forbidden transition, due to its strong spin–orbit coupling associated with the presence of a heavy atom effect. We thus surmised that it could be used for the direct sensitization of low-lying triplet states, in particular for the generation of urethanes via decarboxylation of oxamic acids.
 | ||
| Fig. 1 Decarboxylation of oxamic acids under NIR conditions. | ||
During the course of the present work, Rovis et al. made use of this catalyst in a series of photocatalyzed transformations,8 which prompt us to report our own investigations on the osmium complex-mediated decarboxylation of oxamic acids under NIR irradiation. We describe here our survey of conditions using this photocatalyst under a variety of conditions to initiate the decarboxylation of oxamic acids in the presence of alcohols and heterocycles to produce respectively urethanes and amides under mild conditions (Fig. 1). Penetration depth experiments illustrate the advantages of NIR irradiation.
Optimization studies were performed using oxamic acid 1a, BI-AOc as the hypervalent iodine oxidant and EtOH 2a as a model alcohol.9 TTA-UC conditions were first attempted using Os(bptpy)2(PF6)2 (0.2 mol%) and a perylene derivative (TTBP) as sensitizer/annihilator pair, in the presence of Ru(bpy)3Cl2 as a photocatalyst (PC). Irradiation in the red region (660 nm) led to the desired urethane 3a in 82% isolated yield (Table 1, entry 1).
|
|
||||
|---|---|---|---|---|
| Entrya | Photocat: Sens/An/PC | Oxidant | Time (h) | Yieldb (%) |
| a Unless otherwise stated, all reactions were performed with 1a (0.25 mmol), 2a (3.0 eq.), Os(bptpy)2(PF6)27a,8 (0.2 mol%), TTBP (3 mol%), PC: VL photocatalyst (1.0 mol%) in DCE (0.1 M). Irradiation at 660 nm. b Yields of 3a determined by 1H NMR with 1,3,5-trimethylbenzene as an external standard (isolated yields of 3a in brackets). c AcrMes+ClO4− (2.0 mol%) was used. d Reaction without light. e Reaction performed using 780 nm NIR. f Reaction performed using blue LED (455 nm). g Reaction using 1.0 eq. of BI-OAc. h Reaction performed in CH3CN. | ||||
| 1 | Os(II)/TTBP/Ru(bpy)3Cl2 | BI-OAc | 24 | 88 (82) |
| 2 | Os(II)/TTBP/4-CzIPN | BI-OAc | 24 | 42 |
| 3c | Os(II)/TTBP/AcrMes+ClO4− | BI-OAc | 24 | 80 |
| 4 | Os(II/TTBP/Ru(bpy)3Cl2 | PIDA | 24 | 64 |
| 5 | Os(II)/AcrMes+ClO4− | BI-OAc | 24 | 77 |
| 6 | Os(II)/Ru(bpy)3Cl2 | BI-OAc | 24 | 78 |
| 7 | Ru(bpy)3Cl2 | BI-OAc | 24 | 9 |
| 8 | TTBP | BI-OAc | 24 | ND |
| 9 | — | BI-OAc | 24 | ND |
| 10 | Os(II) (0.2) | BI-OAc | 24 | 52 |
| 11 | Os(II) (0.3) | BI-OAc | 24 | 93 (87) |
| 12d | Os(II) | BI-OAc | 24 | ND |
| 13 | Os(II) | — | 24 | ND |
| 14e | Os(II) (0.3) | BI-OAc | 24 | 60 |
| 15f | Os(II) | BI-OAc | 24 | 73 |
| 16g | Os(II) (0.3) | BI-OAc | 24 | 89 |
| 17 | Os(II) (0.3) | BI-OAc | 6 | 90 |
| 18h | Os(II) (0.3) | BI-OAc | 6 | 45 |

Changing the PC for more benign organophotocatalysts led to a lower yield with 4-CzIPN (entry 2), and a similar one for acridinium salt (entry 3).9 The iodine source was also varied, but PIDA turned out to be less efficient than BI-OAC as already observed using blue light irradiation (entry 4).9 Surprisingly, when the reaction was carried out without the annihilator (entry 5), the yield remained the same as that in entry 3. A similar behaviour was observed for Ru(bpy)3Cl2 conditions in Entry 6 versus 4, confirming that the annihilator had no effect on the reaction. However, in the absence of the Sen/An pair, the photocatalyst Ru(bpy)3Cl2 led to 3a in ∼9% indicating that the Os(II) sensitizer is crucial (entry 7). This was further confirmed when both Os(II) and Ru catalyst or when all photoactive species were removed (entry 8 and 9). Using the Os(II) sensitizer alone finally afforded 3a, albeit in modest yield (entry 10), in agreement with recent observations by Rovis and co-workers.8 Optimal conditions were however reached by slightly increasing the amount of Os(II) to 0.3 mol% (entry 11). Reaction in the absence of light (entry 12) or oxidant (entry 13) led to no product, confirming that the reaction was photocatalyzed by the Os(II) complex under NIR irradiation. With a longer wavelength (λmax = 780 nm), the procedure still delivered 3a in moderate yield (entry 14). In this case, although Os(bptpy)2(PF6)2 absorbs efficiently (Fig. S2, ESI†), the emission spectrum of the lamp decreases in the NIR region (Fig. S4, ESI†). With blue LEDs (450 nm), 3a was also observed in a satisfying yield (entry 15) due to the Os(II) ter-pyridine complex being panchromatic10 and absorbing strongly over the visible spectrum, while generating the same excited state.7a,8 Reducing the amount of BI-OAc had little effect on the yield (entry 16). Decreasing the reaction time to 6 h still led to high conversion (entry 17). Finally, the reaction did not work efficiently with CH3CN as a solvent (entry 18).
Under the optimal conditions (Table 1, entry 11), the substrate scope was then extended varying both the nature of oxamic acids 1 and that of alcohols 2 (Scheme 1). Yields are generally high, in the range of those observed using VL irradiation, indicating the efficiency of NIR and the Os(II) catalyst. The process is compatible with a range of alcohols including diols, fluorinated, chlorinated and unsaturated alcohols. Secondary alcohols react even at room temperature, as indicated by the formation of 3p, in contrast with standard reactions between isocyanates and alcohols which require catalysts.11 Variation is allowed on the oxamic acid moiety, including heterocycles such as furan and thiophene 3c-d. Electron-poor arenes (3e, 3j) are also compatible with these reaction conditions. The reaction was performed on enantiomerically pure oxamic acids leading to the corresponding urethanes 3o-q with no erosion of enantiomeric purity. Bis-oxamic acids were finally shown to afford bis-urethanes 3r-s in good yields.
 | ||
| Scheme 1 Os(II)-mediated decarboxylation of oxamic acids in the presence of alcohols 2 under NIR irradiation. aReaction time of 6 h. | ||
The Os(II)-mediated photocatalyzed decarboxylation under NIR light was also extended to the addition of carbamoyl radicals onto heteroarenes.9b These “Minisci” additions proved to be particularly efficient, proceeding overnight to afford amides 5a-i in generally high yields with excellent regioselectivities in good agreement with our previous report using visible light (Scheme 2).9b Again, addition of homochiral oxamic acid 1o onto heterocycle 4c occurred cleanly to afford 5b with no loss of enantiopurity. Two consecutive additions were observed during reaction with phthalazine to provide the bis-amide 5h in 76% yield.
 | ||
| Scheme 2 Os(II)-mediated decarboxylation of oxamic acids in the presence of heteroarenes under NIR irradiation. aReaction time of 6 h. bReaction time of 24 h. | ||
The study was continued by investigating the penetration depth of the light used for exciting the Os(II) complex using various barriers between the light source and the reaction medium (Fig. S9, ESI†).8 Reaction of oxamic acid 1a with EtOH 2a using the Os(II) catalyst behind a wall of paraffin wax (1.5 cm) or pig skin (1 cm) was thus shown to afford 3a respectively with 74% and 89% yield at 660 nm, but only traces at 455 nm (to be compared with entry 15, Table 1). Further studies summarized in Table 2 also compared the present protocol using Os(II)-LED at 660 nm with that of the visible light process developed earlier using 4-CzIPN at 455 nm.9a A slight decrease in yield was thus apparent with VL using a paraffin wax of 1 mm (Table 2, entry 2). Only 7 mm of this film prevented VL from promoting the reaction, while the yield using NIR was hardly modified (entry 3). Increasing the paraffin film thickness to 1.5 cm resulted in only a 20% decrease in yield using the NIR process (entry 4), whereas a sheet of white paper or pig skin (ESI†) almost completely stops VL (entries 5 and 6). Finally, when the reaction was performed within a model haemoglobin solution (a porphyrin derivative absorbing strongly in the visible region), blue light penetration was completely blocked, and not even a trace of urethane 3a was observed (entry 7). These few experiments thus demonstrate unambiguously that low energy NIR irradiation offers an attractive alternative to VL for photochemical activation in viscous systems, for instance in polymer or biological media.4
|
|
|||
|---|---|---|---|
| Entrya | Barrier | (%) 3ab (660 nm) | (%) 3ab (455 nm) |
| a Unless otherwise stated, all reactions were performed: NIR experiment: 1a (0.25 mmol), 2a (3.0 eq.), Os(bptpy)2(PF6)2 (0.3 mol%), LED (660 nm). Visible light experiment: 4-CzIPN (2.0 mol%), LED (455 nm). b Yields of 3a determined by 1H NMR with 1,3,5-trimethylbenzene as an external standard. | |||
| 1 | None | 93 | 94 |
| 2 | Paraffin wax (1 mm) | 92 | 89 |
| 3 | Paraffin wax (7 mm) | 85 | Trace |
| 4 | Paraffin wax (1.5 cm) | 71 | trace |
| 5 | White paper (3 sheets) | 80 | 12 |
| 6 | Pig skin (1 cm) | 86 | 12 |
| 7 | Haemoglobin | 87 | — |
Mechanistic experiments to identify the photogenerated intermediates were carried out. The in situ formation of an isocyanate was first demonstrated by generating isocyanate 6 (1H NMR and FT-IR) as in Scheme 1 without alcohols (Scheme 3). Previous trapping experiments showed that a carbamoyl radical was generated during the VL-mediated process.9 Under Os(II)-NIR conditions, oxamic acid 1a similarly generated a carbamoyl radical which was trapped by TEMPO, leading to amide 7, albeit in trace amount. The excited-state reactivity of the Os photocatalyst was investigated by luminescence quenching experiments (Table 3 and Fig. S14, S15, ESI†). These show that only the combination of 1a and BI-OAc substantially quenches the emission of the excited osmium species, in agreement with a mechanism in which a reaction between 1a and BI-OAc generates an intermediate capable of efficiently quenching the excited photocatalyst. In contrast, 1a alone has no effect on the overall emission quantum yield even though a reduction in the average luminescence lifetime is observed.
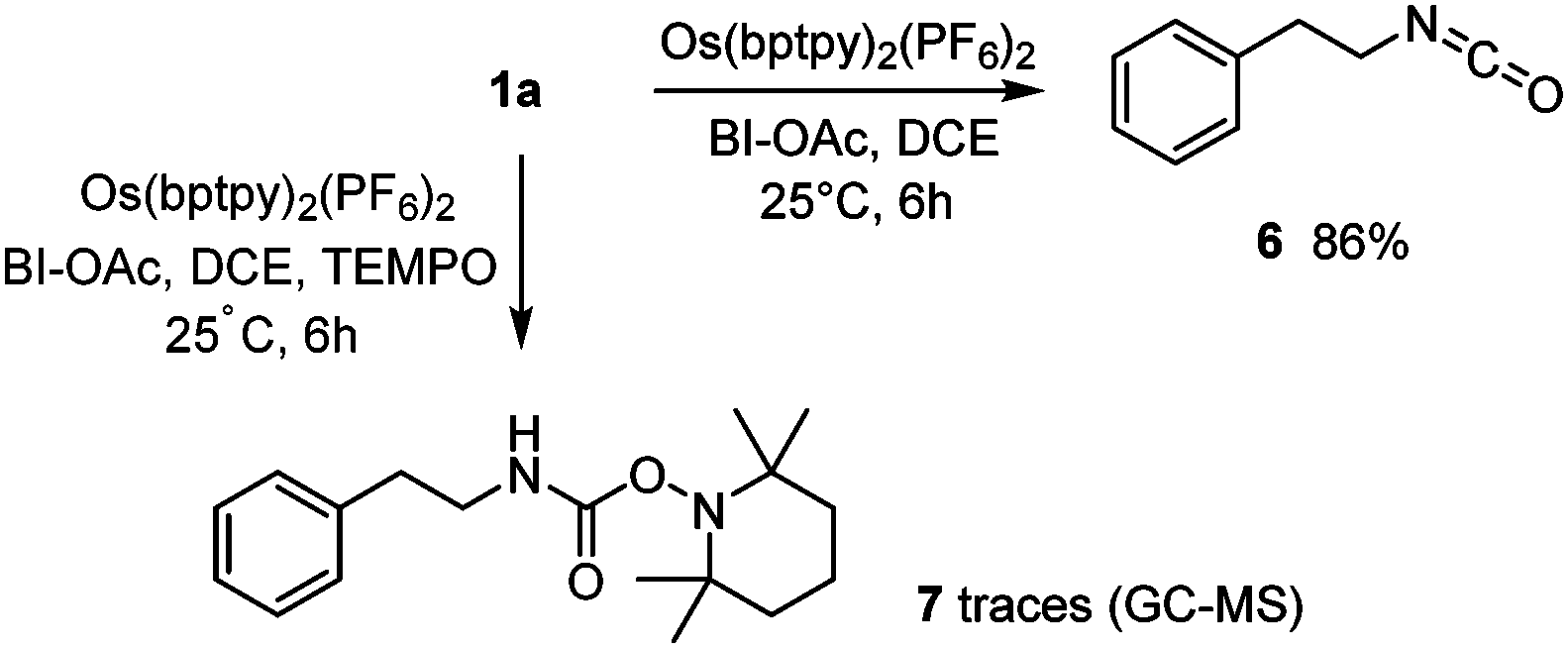 | ||
| Scheme 3 Trapping experiments. | ||
| Quencherc | λ em | Φ em(rel)de | Luminescence decay (ns)b | |
|---|---|---|---|---|
| τ 1 | τ 2 | |||
| a 250 μM in deaerated dichloromethane/acetonitrile (20% v/v) solutions. b λ ex = 410 nm, 600 nm long-pass filter. Relative weighted contribution given in parenthesis. An additional minor short-lived component (τ ≤ 3 ns) is present in all samples. c 50 mM concentration in each species. d λ ex = 450 nm. e Relative intensity. | ||||
| None | 736 | 1.00 | 80 (35) | 214 (58) |
| 1a | 736 | 1.03 | 11 (54) | 141 (31) |
| BI-OAc | 736 | 1.24 | 53 (18) | 285 (72) |
| 1a + BI-OAc | 737 | 0.70 | 8.6 (52) | 122 (34) |
The substantial increase in emission intensity upon addition of BI-OAc is intriguing (Fig. S13, ESI†). It is accompanied by an increase of the average lifetime but no change in the emission spectrum, implying that a deactivation pathway intrinsic to the excited Os complex is inhibited. 1H NMR studies evidence that the addition of BI-OAc results in the disappearance of the ter-pyridine 6,6′′ protons, indicating that they are likely in the vicinity of the hypervalent iodine (Fig. S10–S12, ESI†). The existence of aromatic π-stacking interactions between extended ter-pyridine complexes of Os(II)12 suggests that the BI-OAc may be weakly bound to the photocatalyst, thereby reducing its non-radiative deactivation through rigidification of the coordination sphere.13 Increasing the excited state lifetime of the photocatalyst can be important in cases when, like here, the quencher may be present at low concentrations.
A tentative mechanism for the Os(bptpy)2(PF6)2–mediated decarboxylation of oxamic acids is depicted in Fig. 2. As previously shown,9 the reaction likely proceeds through the formation of an hypoiodite such as I, whose reduction by photoexcited Os(II) leads to radical-anion II (EOs(II)*/Os(III) = −0.67 V).8 Mesolytic cleavage of the latter then generates carbamoyl radical III after decarboxylation. III may then be oxidized by Os(III) (EOs(II)/Os(III) = +1.08 V)8 into a protonated isocyanate IV, which can react with an alcohol to afford the expected carbamate, concomitantly returning the Os catalyst to its initial oxidation state. Alternatively, in the presence of heterocycles, III can add onto the protonated aromatic system to afford IV′, the oxidation of which into V′ and rearomatization afford the desired amide.
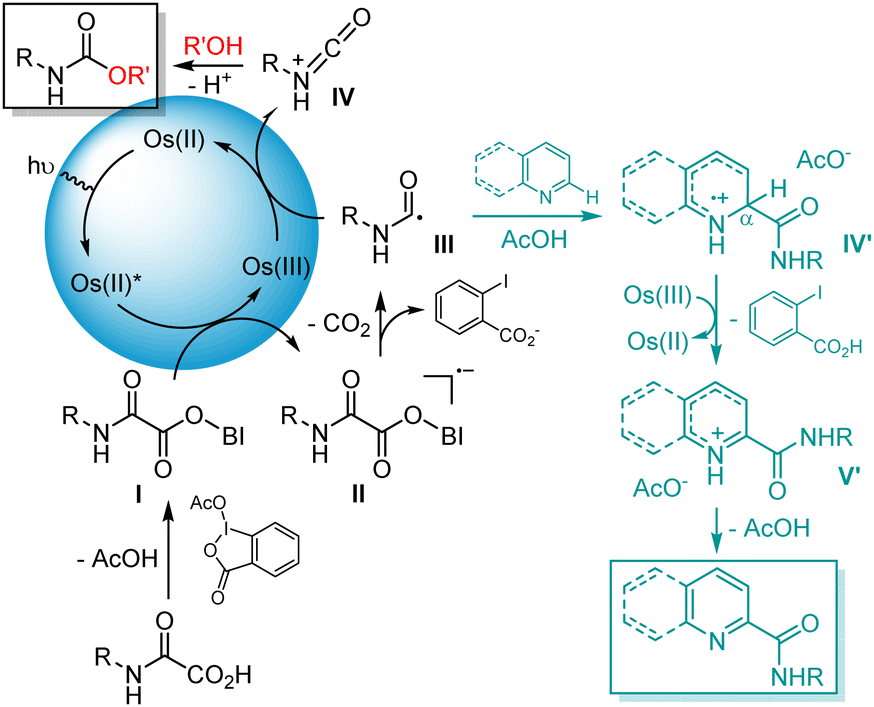 | ||
| Fig. 2 Mechanism of the Os(II)-mediated decarboxylation of oxamic acids in the presence of alcohols or heteroarenes under NIR irradiation. | ||
In summary, we report the photocatalyzed decarboxylation of oxamic acids under NIR conditions to access urethanes and heterocyclic amides using very low amounts (0.3 mol%) of an Os(II) complex. These conditions allow the decarboxylation to be performed behind various polymer or biological barriers, which should find applications on large scale processes or in heterogeneous or viscous media where visible light is not operational.4
IMO thanks the Alex-Ekwueme Federal University Ndufu-Alike Ikwo (AE-FUNAI) for a PhD grant. We are grateful to the ANR (NCO-INNOV, No. 20-CE07-0015-01), the University of Bordeaux and the CNRS for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343 CrossRef CAS PubMed; (b) Z. Li, X. Zou, F. Shi, R. Liu and Y. Yagci, Nat. Commun., 2019, 10, 3560 CrossRef PubMed; (c) Z. Wu, K. Jung and C. Boyer, Angew. Chem., Int. Ed., 2020, 59, 2013–2017 CrossRef CAS PubMed; (d) L. Mei, J. M. Veleta and T. L. Gianetti, J. Am. Chem. Soc., 2020, 142, 12056 CrossRef CAS PubMed; (e) A. R. O. Kosso, N. Sellet, A. Baralle, M. Cormier and J.-P. Goddard, Chem. Sci., 2021, 12, 6964 RSC; (f) A. H. Bonardi, F. Dumur, T. M. Grant, G. Noirbent, D. Gigmes, B. H. Lessard, J.-P. Fouassier and J. Lalevée, Macromolecules, 2018, 51, 1314 CrossRef CAS.
- For reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (c) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034 CrossRef CAS PubMed.
- (a) L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730 CrossRef CAS PubMed; (b) C. C. Le, M. K. Wismer, Z.-C. Shi, R. Zhang, D. V. Conway, G. Li, P. Vachal, I. W. Davies and D. W. C. MacMillan, ACS Cent. Sci., 2017, 3, 647 CrossRef CAS PubMed.
- (a) S. Daehne and U. Resch-Genger, Near-Infrared Dyes for High Technology Applications, ed., O. S. Wolfbeis, Springer, Netherlands, Dordrecht, 1998 CrossRef; (b) A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710 CrossRef CAS PubMed; (c) S. Shanmugam, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55, 1036 CrossRef CAS PubMed.
- (a) T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560 CrossRef CAS; (b) J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395 CrossRef CAS PubMed.
- (a) M. P. Rauch and R. R. Knowles, Chimia, 2018, 72, 501 CrossRef CAS PubMed; (b) N. Sellet, M. Cormier and J. P. Goddard, Org. Chem. Front., 2021, 8, 6783 RSC; (c) L. Huang, W. Wu, Y. Li, K. Huang, L. Zeng, W. Lin and G. Han, J. Am. Chem. Soc., 2020, 142, 18460 CrossRef CAS.
- (a) Y. Sasakia, S. Amemori, H. Kouno, N. Yanai and N. Kimizuka, J. Mater. Chem. C, 2017, 5, 5063 RSC; (b) N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2020, 59, 10252 CrossRef CAS.
- B. D. Ravetz, N. E. S. Tay, C. L. Joe, M. Sezen-Edmonds, M. A. Schmidt, Y. Tan, J. M. Janey, M. D. Eastgate and T. Rovis, ACS Cent. Sci., 2020, 6, 2053 CrossRef CAS PubMed.
- (a) G. G. Pawar, F. Robert, E. Grau, H. Cramail and Y. Landais, Chem. Commun., 2018, 54, 9337 RSC; (b) A. H. Jatoi, G. G. Pawar, F. Robert and Y. Landais, Chem. Commun., 2019, 55, 466 RSC.
- M. Irikura, Y. Tamaki and O. Ishitani, Chem. Sci., 2021, 12, 13888–13896 RSC.
- (a) J. Alsarraf, Y. Ait Ammar, F. Robert, E. Cloutet, H. Cramail and Y. Landais, Macromolecules, 2012, 45, 2249 CrossRef CAS; (b) H. Sardon, A. C. Engler, J. M. W. Chan, J. M. García, D. J. Coady, A. Pascual, D. Mecerreyes, G. O. Jones, J. E. Rice, H. S. W. Horn and J. S. L. Hedrick, J. Am. Chem. Soc., 2013, 135, 16235 CrossRef CAS PubMed.
- D. Gut, A. Rudi, J. Kopilov, I. Goldberg and M. Kol, J. Am. Chem. Soc., 2002, 124, 5449 CrossRef CAS PubMed.
- P. P. Lainé, S. Campagna and F. Loiseau, Coord. Chem. Rev., 2008, 252, 2552 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc03155h |
| This journal is © The Royal Society of Chemistry 2022 |