
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand-based drug repurposing strategy identified SARS-CoV-2 RNA G-quadruplex binders†
Federica
Moraca
 ab,
Simona
Marzano
a,
Francesco
D'Amico
c,
Antonio
Lupia
ab,
Silvia
Di Fonzo
c,
Eleonora
Vertecchi
d,
Erica
Salvati
d,
Anna
Di Porzio
a,
Bruno
Catalanotti
a,
Antonio
Randazzo
a,
Bruno
Pagano
*a and
Jussara
Amato
*a
ab,
Simona
Marzano
a,
Francesco
D'Amico
c,
Antonio
Lupia
ab,
Silvia
Di Fonzo
c,
Eleonora
Vertecchi
d,
Erica
Salvati
d,
Anna
Di Porzio
a,
Bruno
Catalanotti
a,
Antonio
Randazzo
a,
Bruno
Pagano
*a and
Jussara
Amato
*a
aDepartment of Pharmacy, University of Naples Federico II, Via D. Montesano 49, 80131 Naples, Italy. E-mail: bruno.pagano@unina.it; jussara.amato@unina.it
bNet4Science srl, University “Magna Græcia” of Catanzaro, 88100 Catanzaro, Italy
cElettra-Sincrotrone Trieste S. C. p. A., Science Park, 34149 Trieste, Italy
dInstitute of Molecular Biology and Pathology, National Research Council, Via degli Apuli 4, 00185 Rome, Italy
First published on 27th September 2022
Abstract
The single-stranded RNA genome of SARS-CoV-2 contains some G-quadruplex-forming G-rich elements which are putative drug targets. Here, we performed a ligand-based pharmacophore virtual screening of FDA approved drugs to find candidates targeting such RNA structures. Further in silico and in vitro assays identified three drugs as emerging SARS-CoV-2 RNA G-quadruplex binders.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the highly contagious virus responsible for the ongoing COVID-19 pandemic.1 Although the SARS-CoV-2 vaccination campaign is showing its positive effect,2,3 this pandemic remains a global emergency and the search for drugs to treat the viral infection is still an urgent task. However, traditional drug discovery is a long-cycle process with low success rates.4
In this context, repurposing of already approved drugs is currently one of the most attractive propositions because it involves the use of de-risked compounds, with potentially lower development costs and shorter development timelines.4,5 A widely used approach to drug repurposing starts with virtual screening (VS) of existing drugs employing computational methods which are fast, low-cost screening processes. Many drugs are already being successfully repurposed to treat various diseases, including viral infections.5 Thus, with the aim of boosting the arsenal against COVID-19, scientists also began to explore repurposed therapeutic molecules.6–8
So far, almost all new antiviral therapeutic strategies focus on targeting proteins.8,9 However, the threat posed by SARS-CoV-2 infection requires exploring also plausible alternative approaches, such as targeting viral RNA and, in particular, its secondary structures.10,11 Indeed, the folding of specific regions of the viral genomic RNA into certain secondary structures may hinder the viral genome expression and replication by acting as roadblocks for viral RNA transcription and/or as hallmarks for the attachment of RNA processing machinery.
Among these structures are the G-quadruplexes (G4s), four-stranded structures that can be formed by the folding on itself of single-stranded guanine-rich DNA or RNA sequences.12a,b A G4 structure is characterized by the stacking of two or more planar arrangements of four guanines (G-tetrads) stabilized by Hoogsteen hydrogen bonds and cation coordination. These structures may occur in sequences with at least four contiguous tracts of two or more guanines interspersed with sequences forming the so-called loops.
Critical roles for G4s have been described in several viruses,13,14 including single-stranded RNA viruses, and some G4-targeting compounds have shown antiviral activity,15 thus suggesting G4 specific compounds as potential antiviral agents. Recent reports have identified a number of putative G4-forming sequences in the genome of SARS-CoV-2, and some of them were demonstrated to form G4s in vitro.11,16–18
Here, by using ligand-based VS (LBVS) of FDA approved drugs and multiple biophysical techniques, we identified therapeutic molecules able to bind and stabilize SARS-CoV-2 G4-forming RNA and also provided a plausible mechanism of action of such molecules at the molecular level.
Molecules with similar structures tend to have similar properties and functions. Therefore, in LBVS, a pharmacophore model could be derived to define the structural features required to bind a target and exert biological activity.19 Following this approach, drugs matching the structural and geometrical features of typical G4 binders should have analogous G4-binding properties. Thus, starting from known active RNA G4 ligands from the literature (training set, Table S1, ESI†), 3D pharmacophore models were generated and validated before performing the drug repurposing LBVS (for details on training set generation, see the Experimental section, ESI†). 3D ligand-based pharmacophores were built on the 2D Fingerprint clustered training set, generating further 50 conformers to increase the chances that ligands matched the pharmacophores of the known G4 binders. A library of decoys was used as a benchmark test set to validate the predictive power of each hypothesis, leading to five best performing pharmacophore models (Fig. 1). All five models showed some typical features of G4 binders, namely three aromatic rings and a hydrogen bond donor or a positive ionisable group, which however are placed in different 3D spatial arrangements, as highlighted by their distances and by the different directionality of the vectors D (Fig. 1F–J), allowing us to retrieve different RNA G4 binders from the training set (Table S2, ESI†). Thus, the five pharmacophore models were employed to screen a library of ∼3000 FDA-approved drugs, considering the fitness score of the training set as a cut-off value to choose the best-matching drugs. The best hits, along with their original use, are listed in Table S3 (ESI†). Of note, 5 of the 15 putative G4 ligands (Table S4, ESI†) have already been evaluated as possible anti COVID-19 agents.
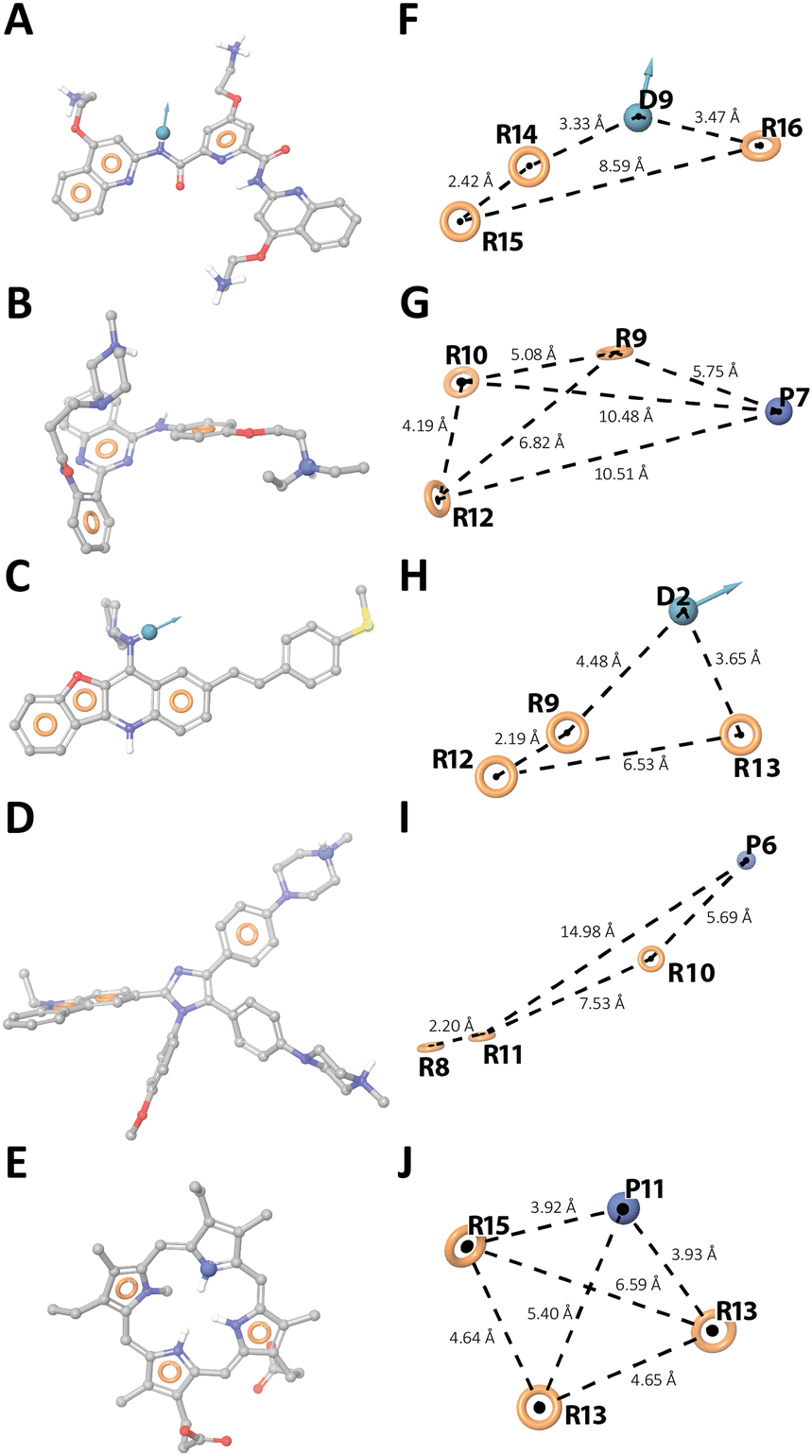 | ||
| Fig. 1 Ligand-based 3D pharmacophore models. (A–E) Hy1, Hy3, Hy5, Hy8, and Hy9 overlapping the reference ligands PDP, 19, 4a-10, IZCZ-3, and NMM-IX, respectively (Table S1, ESI†). (F–J) Pharmacophore hypotheses and distances between the pharmacophoric sites (dashed lines). Aromatic rings, H-bond donor, and positive ionizable groups are labelled as R, D, and P, respectively. The tolerance radius of each feature was set to 2 Å. | ||
To investigate their G4-binding properties, we employed three biologically relevant G-rich sequences of SARS-CoV-2 (G4-1, G4-2, and G4-3, ESI†) found in the coding sequence regions of nucleocapsid protein, non-structural protein 10, and non-structural protein 3, as potential targets. Such sequences have already been shown to form stable G4s.11,18 First, the ability of the drugs to bind those G4s was assessed by fluorescent intercalator displacement (G4-FID) assay, which relies on a light-up fluorescent probe (thiazole orange, TO) that binds to the G4 structures and can be competitively displaced by candidate ligands, thus enabling the determination of their relative affinity.20 Pyridostatin (PDS), a well-known G4 binder, was used as a positive control. The results of G4-FID assay (Fig. S1 and Table S5, ESI†) showed that only Netarsudil and Quercetin (along with PDS) were able to effectively displace TO from all three G4 structures. Pranlukast turned out to be a very good TO competitor in the case of G4-1, while Ledipasvir and Osimertinib exhibited a certain degree of TO displacement only from G4-1 and G4-3, respectively. Based on DC50 values, the drugs were ranked as follows: (i) drugs with low to null affinity for G4s (DC50 > 10.0 μM), (ii) drugs with moderate affinity (5.0 < DC50 < 10.0 μM) (Ledipasvir for G4-1, Osimertinib and Quercetin for G4-3), and (iii) good G4 binders (DC50 < 5.0 μM) (Netarsudil, Pranlukast for G4-1, Quercetin for G4-1 and G4-2).
To study the G4-stabilizing effect of ligands showing at least moderate affinity for those targets, circular dichroism (CD) melting experiments were performed. First, the structures adopted by G4-1, G4-2 and G4-3 were verified by recording the relative CD spectra, which showed a maximum around 266 nm and a minimum at 240 nm (Fig. S2, ESI†) indicating the presence of parallel-stranded G4s. Melting and annealing profiles of G4s (recorded at 1.0 and 0.5 °C min−1) were then collected by following the changes in CD signal at 266 nm (Fig. S3, ESI†). As for G4-1, melting and annealing curves were superimposable, showing that the unfolding and folding processes are at thermodynamic equilibrium and the Tm can be accurately determined. Conversely, hysteresis was observed for G4-2 and G4-3, indicating that they are affected by the kinetics of the process (which prevents reproducible measurements) and suggesting the presence of intermolecular G4 species or higher order structures through self-association between G4 units.18 Therefore, in subsequent studies, we decided to focus only on G4-1. The selected drugs significantly increased the thermal stability of G4-1 (Fig. S4, ESI†). Interestingly, the best effects were found for drugs that showed higher affinity for G4-1 in the G4-FID assay (Netarsudil, Pranlukast, and Quercetin). Upon interacting with Ledipasvir, Osimertinib, and Quercetin, no significant variations in the CD spectrum of G4-1 were detected (Fig. S4, ESI†), implying that it kept its parallel G4 structure. Conversely, Netarsudil and Pranlukast seem to alter the native conformation of G4-1. Actually, these two drugs exhibit CD signal in the wavelength region of G4 (Fig. S5, ESI†). However, at the wavelength at which the melting experiments were recorded, the signal of both is close to zero, thus not significantly affecting the results. On the other hand, the experimental spectra of the two G4/drug mixtures differ sizeably from the corresponding spectra resulting from the arithmetic sum of the single spectra (Fig. S6, ESI†), highlighting once again their interaction.
To evaluate the affinity of Netarsudil, Pranlukast, and Quercetin for G4-1, fluorescence titration experiments were performed. Fluorescence emission spectra of drugs in the absence and presence of increasing amounts of G4 were recorded (Fig. S7, ESI†). On addition of RNA, fluorescence quenching and enhancement were observed for Netarsudil and Quercetin, respectively. Conversely, no relevant change in fluorescence intensity was observed for the intrinsically weakly fluorescent Pranlukast, not allowing us to quantify its affinity. Thus, binding isotherms were obtained by plotting fluorescence changes with G4 concentration (Fig. S7, ESI†), and the curves were fitted giving dissociation constants (Kd) of 0.8 (±0.2) and 13 (±5) μM for Netarsudil and Quercetin, respectively.
To get some information on the selectivity of the drugs, we evaluated their ability to stabilize the G4s derived from the 5′ untranslated region of BCL-2 (BCL2-G4) and the long non-coding RNA GSEC (GSEC-G4) (Fig. S8 and S9, ESI†). CD melting data show that they do not significantly increase the thermal stability of GSEC-G4 (ΔTm ≤ 2 °C). Conversely, while Netarsudil stabilizes BCL2-G4 marginally (ΔTm = 2.6 °C), Pranlukast and Quercetin show a moderate stabilizing capacity (ΔTm of 5.3 and 5.4 °C, respectively), even if lower than that found for G4-1.
UV resonance Raman (UVRR) spectroscopy was then employed to get insights into the binding mode of drugs to G4-1.21 UVRR spectra of G4-1, ligands, and corresponding complexes, all recorded at 266 nm and processed following standard procedures,21 are shown in Fig. 2. In each panel of Fig. 2, the difference between the spectrum of the complex and that corresponding to the arithmetic sum of constituents (see ESI†) is shown to emphasize the spectral perturbations induced by the interaction. A change in the intensity and/or position of the bands (Fig. S10–S13, ESI†) indicates that an interaction is occurring and suggests the structural moieties involved. This analysis is not straightforward for Netarsudil, as most of its bands overlap those of RNA, precluding the possibility of obtaining clear information on the drug-binding regions of RNA. The only exceptions are the spectral variations at 1334, 1480, 1510, and 1579 cm−1, which are associated to the adenine ring vibrations. Compared to the sum, the experimental spectrum of the RNA/Netarsudil complex shows an intensity increase of these bands, indicating that adenine residues of loop are involved in drug binding. Noteworthy, Netarsudil's strong spectral contribution allowed us to evaluate the parts of the drug involved in the interaction. The positive 1646 and negative 1657 cm−1 peaks clearly indicate a shift of the peak at 1652 cm−1, which is attributed to stretching of the rA and rB rings of the drug (Table S6 and Fig. S11, ESI†). Similarly, the positive 1408 and negative 1417 cm−1 peaks reflect a downshift of the peak at 1414 cm−1, which is mainly associated with the C–C/C–N stretching modes of the rA and rB rings. Conversely, no significant change in the peak at 1612 cm−1 was observed, corresponding to the stretching modes of rC and rD rings of the drug. These data suggest that rA and rB rings should be primarily involved in RNA binding.
 | ||
| Fig. 2 UVRR spectra to study the binding of Netarsudil (N), Pranlukast (P), and Quercetin (Q) to G4-1 RNA. From bottom to top: RNA (blue); drug (green); RNA/drug complex (black); arithmetic sum of RNA and drug spectra (red); normalized difference between the spectra of the complex and the arithmetic sum (diff.). Spectra were normalized with the RNA spectrum intensity. | ||
Pranlukast and Quercetin showed negligible UVRR spectral contributions compared to G4-1, so binding-induced spectral perturbations could be easily correlated with the band changes of nucleotides. Upon RNA/Quercetin interaction, a decrease in intensity of the difference spectrum at 1231, 1293, 1393, 1530 and 1625 cm−1, related to bands of U and C residues (Fig. 2), was observed, suggesting their involvement in the interaction. Noteworthy, the presence of a minimum and a maximum at 1476 and 1493 cm−1, respectively, due to a shift of the band at 1483 cm−1 which is mainly related to guanines,21 indicate that these residues take part in the interaction. This is confirmed by the intensity increase and redshift of the band at 1606 cm−1 which could be due to both changes in the guanine normal modes and a downshift of the Quercetin peak (ring stretching vibration) from 1620 to 1604 cm−1 (Table S7, ESI†), overall suggesting that end-stacking is the favoured binding mode for this drug. As for Pranlukast, the difference spectrum shows changes associated to A, G, and U, although of lower intensity than those observed for Quercetin. Interestingly, a clear variation of the peak at 1642 cm−1 is observed, which corresponds mainly to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O2 and CO4 stretching vibrations of the drug in combination with N1–H and C26–H bending (Table S8, ESI†). Although it is a clear indication that Pranlukast binds to G4-1, this is not enough to obtain precise information on the molecular regions involved in the interaction.
O2 and CO4 stretching vibrations of the drug in combination with N1–H and C26–H bending (Table S8, ESI†). Although it is a clear indication that Pranlukast binds to G4-1, this is not enough to obtain precise information on the molecular regions involved in the interaction.
To elucidate the binding poses of the three drugs to G4-1, docking and molecular dynamics simulations (MDs) were performed. The best docking poses showed that, as suggested by experimental data, Quercetin and Pranlukast preferentially bind to the 5′-end G-tetrad via stacking interactions with G11 and G14 and H-bonds with U4 and C13 (Fig. S14, ESI†). Conversely, Netarsudil was found to interact with the loop on the 3′-end side, in agreement with UVRR results. To further verify the docking results and the geometrical stability of G4-1 upon binding, each complex was submitted to 500 ns of MDs. The most populated poses extracted after MDs are shown in Fig. S15 (ESI†). Root Mean Square Deviation (RMSD) analysis revealed that Quercetin adopts a stable binding pose (Fig. S16A, ESI†), keeping the H-bonds with U4 and C13 for ∼62% of MDs (Table S9, ESI†). As for Pranlukast, despite its chromen moiety is anchored by an H-bond with U4 for ∼82% of MDs (Table S9, ESI†), the flexibility of the phenylbutoxy group induces a slight change in the binding mode (Fig. S16B, ESI†). On the other hand, Netarsudil undergoes a deeper change of its binding mode during MDs (Fig. S16C and Movie S1, ESI†). This is mainly due to the CAAU loop, which fluctuates more than in the other complexes (Fig. S17, ESI†). The binding arrangements of Netarsudil are well highlighted by the analysis of solvent-accessible surface area (Fig. S18, ESI†), which decreases mainly for Netarsudil, A9 and U10 residues following the establishment of π–π stacking interactions between the isoquinoline and the phenyl moieties of the drug and the two RNA residues (Fig. S15C, ESI†). This explains the slightly higher RMSD values for G4-1 in the RNA/Netarsudil complex with respect to the G4-1 free state, contrarily to the Quercetin and Pranlukast effects (Fig. S19, ESI†). Nonetheless, the 2dRMSD matrices show the high tendency of Netarsudil to stabilize the G4-1 (Fig. S20, ESI†) as observed in the other experiments. Overall, these results fully agree with the band changes in the UVRR data induced by ligand binding. Changes in the G4-1 RNA geometry were also analysed by calculating some basic G4 parameters, i.e. the rotation (twist) angle between the two G-tetrads, their planarity, and gyration radii of the G-tetrads and whole G4 (Fig. S21, ESI†).22 No significant changes in the position and planarity of G-tetrads and in G4 structural integrity were observed (Fig. S22–S24, ESI†), again indicating no major structural rearrangements in G4-1.
To evaluate if these drugs could interfere with biological functions such as viral RNA replication, we investigated whether their binding to G4 affected reverse transcription. A synthetic SARS-CoV-2 genomic RNA template was reverse transcribed using specific primers flanking the G4-1 or a non-G4-forming control region in the presence of growing concentrations of drugs. Then, G4-1 or the control region were used for quantitative RT-PCR to assay the levels of reverse transcription. As shown in Fig. 3, the drugs significantly reduced G4-1 reverse transcription compared to untreated samples, when normalized with the control region, thus showing that they may be able to interfere with viral replication.
 | ||
| Fig. 3 Fold change in reverse transcription of the G4-1 region over a non-G4-forming control region from a synthetic SARS-CoV-2 RNA in the presence of the drugs. Untreated conditions were used as a control. | ||
In conclusion, three FDA approved drugs were identified as effective SARS-CoV-2 G4 binders through a ligand-based drug repurposing strategy. Our results lay the basis for further studies aiming to evaluate the antiviral activity of such drugs, while the methodological approach employed will certainly impact medicinal chemistry approaches for targeting of viral RNA G4s, even beyond SARS-CoV-2.
This work was supported by Italian Ministry of University and Research (FISR2020IP_04932 to JA). The authors thank CERIC for the support and facilities [20217014 to JA].
Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Wu, S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Y. Hu, Z.-W. Tao, J.-H. Tian, Y.-Y. Pei, M.-L. Yuan, Y.-L. Zhang, F.-H. Dai, Y. Liu, Q.-M. Wang and J.-J. Zheng, et al. , Nature, 2020, 579, 265–269 CrossRef CAS PubMed.
- E. J. Haas, F. J. Angulo, J. M. McLaughlin, E. Anis, S. R. Singer, F. Khan, N. Brooks, M. Smaja, G. Mircus, K. Pan, J. Southern, D. L. Swerdlow, L. Jodar, Y. Levy and S. Alroy-Preis, Lancet, 2021, 397, 1819–1829 CrossRef CAS.
- K. B. Pouwels, E. Pritchard, P. C. Matthews, N. Stoesser, D. W. Eyre, K.-D. Vihta, T. House, J. Hay, J. I. Bell, J. N. Newton, J. Farrar, D. Crook and D. Cook, et al. , Nat. Med., 2021, 27, 2127–2135 CrossRef CAS PubMed.
- T. T. Ashburn and K. B. Thor, Nat. Rev. Drug Discovery, 2004, 3, 673–683 CrossRef CAS PubMed.
- S. Pushpakom, F. Iorio, P. A. Eyers, K. J. Escott, S. Hopper, A. Wells, A. Doig, T. Guilliams, J. Latimer, C. McNamee, A. Norris, P. Sanseau, D. Cavalla and M. Pirmohamed, Nat. Rev. Drug Discovery, 2019, 18, 41–58 CrossRef CAS PubMed.
- G. Ciliberto and L. Cardone, Drug Discovery Today, 2020, 25, 946–948 CrossRef CAS PubMed.
- Y. L. Ng, C. K. Salim and J. J. H. Chu, Pharmacol. Ther., 2021, 228, 107930 CrossRef CAS PubMed.
- W. D. Jang, S. Jeon, S. Kim and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2024302118 CrossRef CAS.
- O. Minenkova, D. Santapaola, F. M. Milazzo, A. M. Anastasi, G. Battistuzzi, C. Chiapparino, A. Rosi, G. Gritti, G. Borleri, A. Rambaldi and C. Dental, et al. , Mol. Ther., 2022, 30, 1979–1993 CrossRef CAS PubMed.
- S. Sreeramulu, C. Richter, H. Berg, M. A. Wirtz Martin, B. Ceylan, T. Matzel, J. Adam, N. Altincekic, K. Azzaoui and J. K. Bains, et al. , Angew. Chem., Int. Ed., 2021, 60, 19191–19200 CrossRef CAS PubMed.
- C. Zhao, G. Qin, J. Niu, Z. Wang, C. Wang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2021, 60, 432–438 CrossRef CAS.
- (a) G. N. Parkinson, M. P. Lee and S. Neidle, Nature, 2002, 417, 876–880 CrossRef CAS; (b) S. Neidle and G. N. Parkinson, Biochimie, 2008, 90, 1184–1196 CrossRef CAS PubMed.
- E. Lavezzo, M. Berselli, I. Frasson, R. Perrone, G. Palù, A. R. Brazzale, S. N. Richter and S. Toppo, PLoS Comput. Biol., 2018, 14, e1006675 CrossRef PubMed.
- A. Abiri, M. Lavigne, M. Rezaei, S. Nikzad, P. Zare, J.-L. Mergny and H.-R. Rahimi, Pharmacol. Rev., 2021, 73, 897–923 CrossRef CAS.
- E. Ruggiero, I. Zanin, M. Terreri and S. N. Richter, Int. J. Mol. Sci., 2021, 22, 10984 CrossRef CAS PubMed.
- H. Cui and L. Zhang, Front. Microbiol., 2020, 11, 567317 CrossRef PubMed.
- D. Ji, M. Juhas, C. M. Tsang, C. K. Kwok, Y. Li and Y. Zhang, Briefings Bioinf., 2021, 22, 1150–1160 CrossRef CAS PubMed.
- E. Belmonte-Reche, I. Serrano-Chacón, C. Gonzalez, J. Gallo and M. Bañobre-López, PLoS One, 2021, 16, e0250654 CrossRef CAS PubMed.
- A. Maruca, F. A. Ambrosio, A. Lupia, I. Romeo, R. Rocca, F. Moraca, C. Talarico, D. Bagetta, R. Catalano, G. Costa, A. Artese and S. Alcaro, Phys. Sci. Rev., 2019, 4, 20180113 Search PubMed.
- D. Monchaud and M.-P. Teulade-Fichou, Methods in Molecular Biology, 2010, pp. 257–271 Search PubMed.
- S. Di Fonzo, J. Amato, F. D’Aria, M. Caterino, F. D’Amico, A. Gessini, J. W. Brady, A. Cesàro, B. Pagano and C. Giancola, Phys. Chem. Chem. Phys., 2020, 22, 8128–8140 RSC.
- V. Tsvetkov, G. Pozmogova and A. Varizhuk, J. Biomol. Struct. Dyn., 2016, 34, 705–715 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed Experimental section, Tables S1–S9, Fig. S1–S24 and Movie S1. See DOI: https://doi.org/10.1039/d2cc03135c |
| This journal is © The Royal Society of Chemistry 2022 |