
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ionic encapsulation of a methanol carbonylation catalyst in a microporous metal–organic framework†
Samuel A.
Ivko
 ac,
Tom
Bailey
bc,
Lee
Brammer
c and
Anthony
Haynes
*c
ac,
Tom
Bailey
bc,
Lee
Brammer
c and
Anthony
Haynes
*c
aSchool of Chemistry, University of Birmingham, Birmingham B15 2TT, UK
bSchool of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK
cDepartment of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: a.haynes@sheffield.ac.uk
First published on 12th September 2022
Abstract
The anionic rhodium complex cis-[Rh(CO)2I2]−, active in the Monsanto process for acetic acid production, has been heterogenised via Coulombic interactions in the pores of a UiO-66-type metal–organic framework (MOF). The MOF-supported catalyst is active for the carbonylation of methanol and is recyclable, retaining its framework crystallinity following catalysis. Intermediates in the catalytic cycle observed by IR spectroscopy confirm the same mechanism as the established homogeneous process.
The bulk manufacture of acetic acid is an important industrial process with a demand for approximately 20 million tonnes per annum.1 The majority of this demand is met via the catalytic carbonylation of methanol using homogeneous group 9 transition metal catalysts.2 The rhodium/iodide catalysed Monsanto process was originally developed in the 1960s and produces acetic acid with very high selectivity (>99% based on MeOH). The catalytic cycle (Scheme 1) involves a series of anionic Rh iodocarbonyl complexes, beginning with cis-[Rh(CO)2I2]−. This Rh(I) complex can also participate in a water–gas shift cycle via oxidation to the Rh(III) species, [Rh(CO)2I4]−,3 which is inactive for carbonylation and can degrade to give insoluble rhodium compounds. To maintain high catalytic activity and solubility, a relatively high water concentration (∼10 wt%) is employed to suppress accumulation of inactive [Rh(CO)2I4]−, but this increases the cost of drying the acetic acid product by distillation.4 Homogeneous processes that operate at lower water concentration have been developed, including the Celanese AO Plus technology which employs a lithium iodide additive for the Rh catalyst,5 and the iridium-based Cativa process developed by BP.6,7
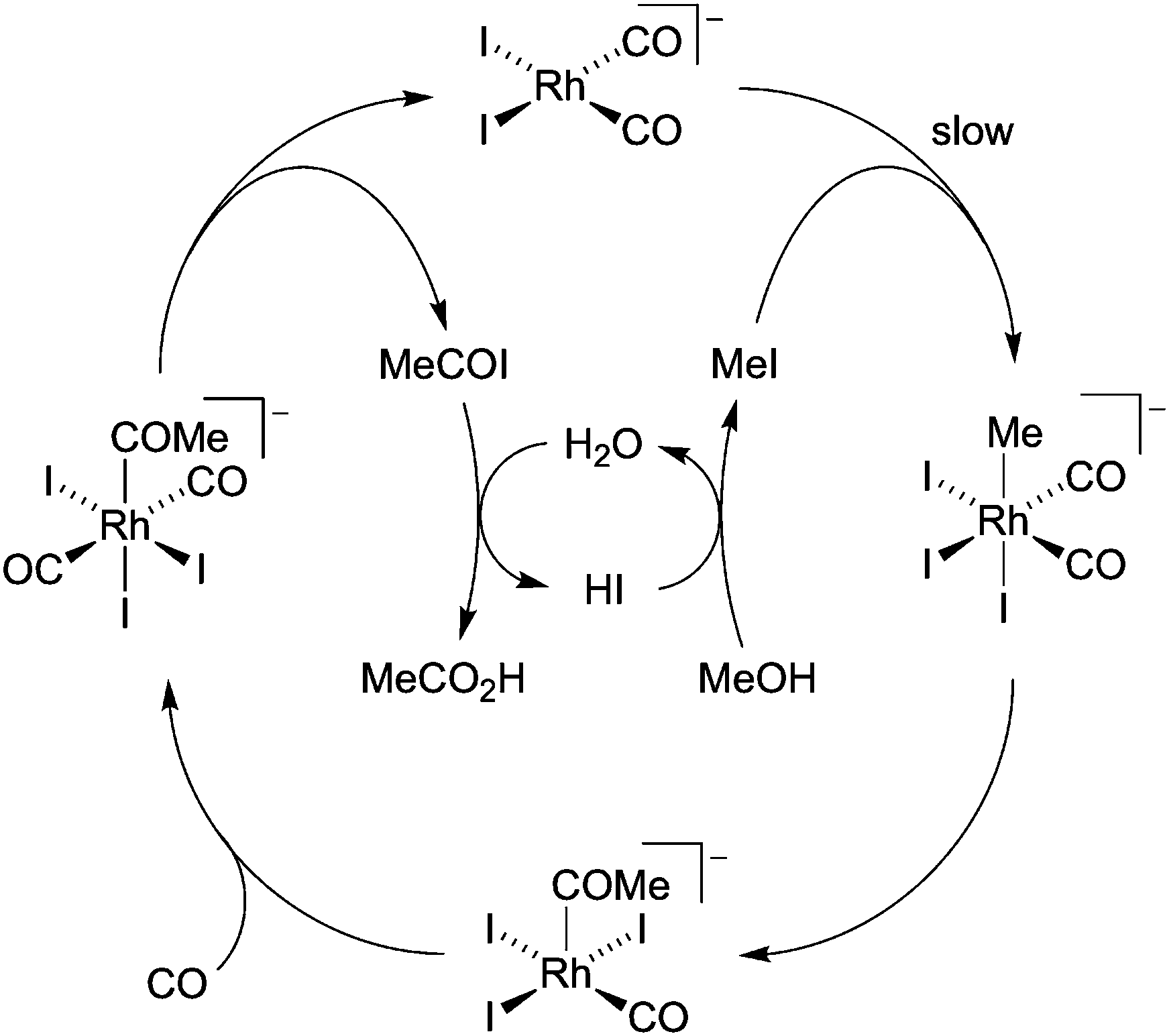 | ||
| Scheme 1 Catalytic cycle for rhodium/iodide catalysed methanol carbonylation. | ||
The issues with catalyst activity and solubility can potentially be alleviated by heterogenisation of the metal complex on a solid support, which constrains the catalyst in the reactor. In the case of an anionic complex such as [Rh(CO)2I2]−, a convenient approach, that maintains the primary coordination sphere of the metal complex, is to employ electrostatic (ion-pair) interactions with a cationic support. This approach was first used by Drago et al. who described the effective immobilisation of [Rh(CO)2I2]− on polymer supports based on N-methylated polyvinylpyridines and found carbonylation activity equal to the homogeneous system with minimal leaching.8 This ionic attachment approach was later adopted in the Acetica™ process developed by Chiyoda/UOP, which attains rates comparable with the homogeneous system but requires lower water concentrations and can employ a higher catalyst loading due to the removal of solubility constraints.9 Mechanistic studies on related polymer-supported systems in our labs in Sheffield,10 including a recent study using dispersible microporous nanoparticles,11 have shown that the same catalytic cycle operates for the supported catalyst as in homogeneous solution phase.
Another class of materials that have received considerable recent attention for their potential as supports for catalyst heterogenisation are metal–organic frameworks (MOFs).12–17 These crystalline, porous materials can incorporate well-defined catalytic sites and facilitate diffusion of reactants and products. The ionic attachment approach has been demonstrated most often with cationic complexes encapsulated in anionic MOFs, for example with Rh and Ir hydrogenation catalysts,18–20 Ru metathesis catalysts21–23 and others.24–27 Alternatively, an anionic complex can be supported in a cationic MOF. For example, exchange of [Co(CO)4]− with halide counter-ions in Cr-MIL-101 gave a supported catalyst that was effective for the carbonylation of epoxides.28,29
In relation to catalytic methanol carbonylation, Burgun et al. used a Mn(II)-based MOF with linkers containing bis(pyrazolyl)methane units to coordinate Rh(CO)2+ moieties. The system showed activity for the carbonylation of MeBr30 and single-crystal crystallographic studies mapped out a series of steps in a catalytic cycle involving N,N-chelated cationic Rh species. Interestingly, one of the materials characterised contained [Rh(CO)2Cl2]− as counter-ion. In this communication, we report the synthesis and characterisation of MOFs having a cationic framework into which the Monsanto carbonylation catalyst [Rh(CO)2I2]− is incorporated, and demonstrate activity for catalytic carbonylation of MeOH/MeI, as well as elucidating steps in the catalytic mechanism.
UiO-type MOFs31 were selected as the platform for heterogenisation of the Rh catalyst. This family of MOFs is based on [Zr6(μ3-O)4(μ3-OH)4]12+ secondary building units (SBUs) with dicarboxylate linkers (e.g. benzene-1,4-dicarboxylate, BDC, in UiO-66). The exceptional thermal and chemical stability resulting from the hard acid-hard base interactions between the highly charged SBUs and carboxylate linkers make this family of MOFs attractive for post-synthetic modification32,33 and catalytic applications.34,35 Liang et al. recently reported a functionalised UiO-66-type MOF, [Zr6O4(OH)4(BDC-Im)6] (BDC-Im = 2-(imidazol-1-yl)benzene-1,4-dicarboxylate), in which N-methylation of the pendant imidazolyl groups on the linkers gave a cationic framework with iodide counter-ions.36 The aim of our study was to incorporate [Rh(CO)2I2]− as the counter-ion into MOFs of this type, enabling catalysis of carbonylation reactions within the confined pore space.
The imidazolyl-functionalised MOF, [Zr6O4(OH)4(BDC-Im)6] was synthesised according to the procedure of Liang et al. (S2.4, ESI†).36 For post-synthetic N-methylation of the imidazolyl groups, we found that higher conversion could be achieved in shorter reactions times by using microwave heating (85%, 1 h) rather than conventional heating (75%, 48 h) as determined by 1H NMR spectroscopy following digestion in D3PO4/D2O (Fig. S5, ESI†). Successful quaternisation was confirmed by solid-state CP-MAS 13C{1H} NMR, in which a new resonance was observed at 38 ppm, consistent with the methyl signal observed in the solution-phase 13C{1H} NMR spectrum of the free quaternised linker, [H2BDC-Im-Me][I] (synthesised independently) (Fig. S6, ESI†).
In order to increase the extent of quaternisation, we sought to reduce steric congestion in the MOF pores by employing a mixed-linker approach, incorporating both BDC and BDC-Im linkers (Fig. 1). A new MOF (1) was synthesised, using a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of H2BDC and H2BDC-Im in the initial reaction mixture. Powder X-ray diffraction (PXRD) confirmed the synthesis of a UiO-66-type MOF as a single phase (Fig. 2a, Pawley fitting in Fig. S7, ESI†). The 1H NMR spectrum of a sample of 1 digested in D2O/NaOD (Fig. S9, ESI†) indicated presence of some modulator-induced acetate present as missing-linker defects in the final product, giving the formula [Zr6O4(OH)4(BDC)4.8(BDC-Im)1.1(OAc)0.1] for 1.
:1 ratio of H2BDC and H2BDC-Im in the initial reaction mixture. Powder X-ray diffraction (PXRD) confirmed the synthesis of a UiO-66-type MOF as a single phase (Fig. 2a, Pawley fitting in Fig. S7, ESI†). The 1H NMR spectrum of a sample of 1 digested in D2O/NaOD (Fig. S9, ESI†) indicated presence of some modulator-induced acetate present as missing-linker defects in the final product, giving the formula [Zr6O4(OH)4(BDC)4.8(BDC-Im)1.1(OAc)0.1] for 1.
 | ||
| Fig. 1 Schematic structure of mixed-linker MOF 1; grey spheres represent [Zr6(μ3-O)4(μ3-OH)4(CO2)12] units, connected by BDC or BDC-Im linkers. | ||
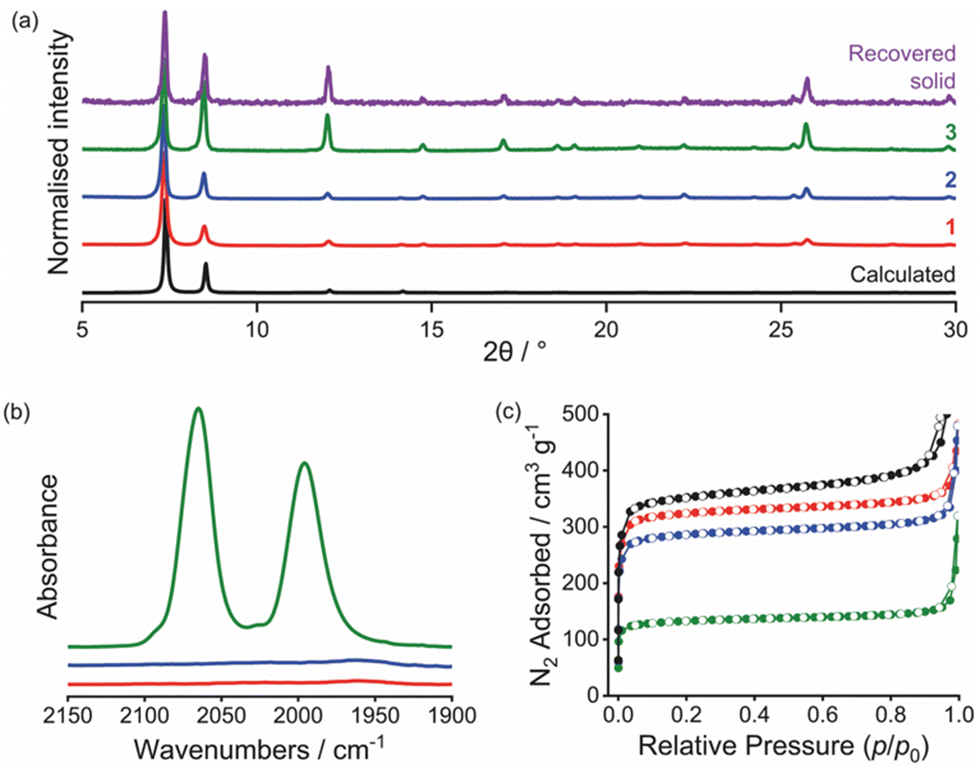 | ||
| Fig. 2 (a) PXRD patterns (including calculated pattern of UiO-66 (black), and pattern of solid recovered after catalysis (purple)), (b) infrared spectra (KBr disc, ν(CO) region), and (c) nitrogen adsorption (filled) and desorption (open) isotherms at 77 K, for UiO-66 (black), 1 (red), 2 (blue) and 3 (green). | ||
Reaction of 1 with MeI yielded [Zr6O4(OH)4(BDC)4.8(BDC-Im-Me)1.1(OAc)0.1][I]1.12 with 98% of the linker imidazolyl sites methylated (S2.7 and Fig. S12, ESI†), demonstrating a significantly higher conversion than for the single-linker MOF36 and PXRD measurements demonstrate that 2 retains crystallinity (Fig. 2a, Pawley fitting in Fig. S10, ESI†). Volumetric N2 gas sorption studies at 77 K (Fig. 2c and Table S1, ESI†) show that the surface area of 1 (1139 m2 g−1) is ∼8% lower than that of the parent UiO-66 (1238 m2 g−1), synthesised in an analogous manner to 1. A further 10% reduction in surface area occurs upon methylation to give 2. The surface areas of 1 and 2, however, are both higher than those reported by Liang et al.36 for the single-linker analogues, [Zr6O4(OH)4(BDC-Im)6] (538 m2 g−1) and [Zr6O4(OH)4(BDC-Im)1.5(BDC-Im-Me)4.5][I]4.5 (328 m2 g−1), indicating that the mixed-linker approach succeeds in reducing pore congestion and facilitating quaternisation of 1 to give 2. The micropore volume also decreases in the order UiO-66 > 1 > 2 (Table S1, ESI†), suggesting that the pendant imidazolyl groups and I− counter-ions occupy the micropores of the MOF.
Using a strategy analogous to that employed previously to generate polymer-supported [Rh(CO)2I2]−,10,11 a sample of 2 was treated with the iodide bridged rhodium dimer [Rh(CO)2I]2, which reacts with the I− counter-ions in 2 (Scheme 2). The presence of a Rh(I) complex cis-[Rh(CO)2I2]− in the product 3 was confirmed by the presence of two ν(CO) absorptions at 2060 and 1986 cm−1 in the IR spectrum (Fig. 2b), very close to the ν(CO) bands for nBu4N[Rh(CO)2I2] in MeOH solution (2060 and 1990 cm−1).37 The PXRD pattern for 3 (Fig. 2a) shows that crystallinity is retained and Pawley fitting (Fig. S13, ESI†) confirms no change in unit cell dimensions (Table S2, ESI†) indicating retention of framework integrity. Changes in relative diffraction intensities between patterns for 2 and 3 (Fig. 2a and Fig. S10, S13, ESI†) are indicative of the change in composition due to replacement of I− anions by [Rh(CO)2I2]− anions within the framework pores. ICP-OES analysis confirmed the presence of rhodium (5 wt%) in the product which corresponds to stoichiometric uptake of Rh relative to the BDC-Im linker, giving the formula [Zr6O4(OH)4(BDC)4.8(BDC-Im-Me)1.1(OAc)0.1][Rh(CO)2I2]1.1 for 3. Volumetric N2 gas sorption measurements showed a significantly smaller surface area and micropore volume for 3, relative to 1 and 2, indicating that the cis-[Rh(CO)2I2]− counter-ions occupy the micropores of the MOF.
 | ||
| Scheme 2 Schematic illustration of post-synthetic quaternisation of 1 to form 2 and subsequent incorporation of Rh(I) complex to give 3. Grey spheres represent [Zr6(μ3-O)4(μ3-OH)4(CO2)12] units. Only the functionalised BDC-Im linker is shown for clarity. (a) 4 M MeI in MeCN, microwave reactor, 120 °C 1 h, (b) [Rh(CO)2I]2 in dry n-hexane, CO atmosphere, RT 16 h. | ||
In a control experiment, reaction of the non-quaternised MOF 1 with [Rh(CO)2I]2 gave a product that exhibited ν(CO) absorptions at 2075 cm−1 and 2007 cm−1 (Fig. S16, ESI†). The higher ν(CO) values indicate formation of a neutral rhodium dicarbonyl species, consistent with coordination of the imidazolyl nitrogen lone pair to a neutral Rh(CO)2I moiety38 (rather than an ion-paired [Rh(CO)2I2]− anion, as in 3).
As observed previously in polymeric systems, MOF-supported cis-[Rh(CO)2I2]− reacts with MeI in an analogous manner to the solution-phase catalyst (Scheme 1 and S2.10, ESI†). After soaking a sample of 3 in MeI overnight, the IR spectrum of the product contained one terminal ν(CO) band at 2060 cm−1, as well as a broad band at around 1735 cm−1 (Fig. 3a). This is consistent with formation of the acetyl complex, [Rh(CO)(COMe)I3]−, resulting from oxidative addition of MeI to cis-[Rh(CO)2I2]−, followed by rapid methyl migration, as observed in the homogeneous system.39 After exposure of the solid product to CO gas (1 atm), the IR spectrum showed absorptions at 2088 and 1694 cm−1 (Fig. 3a), consistent with the dicarbonyl complex trans-[Rh(CO)2(COMe)I3]− also observed in solution.40 Thus, the key organometallic intermediates from the homogeneous catalytic cycle shown in Scheme 1 are also observed in the MOF-supported system.
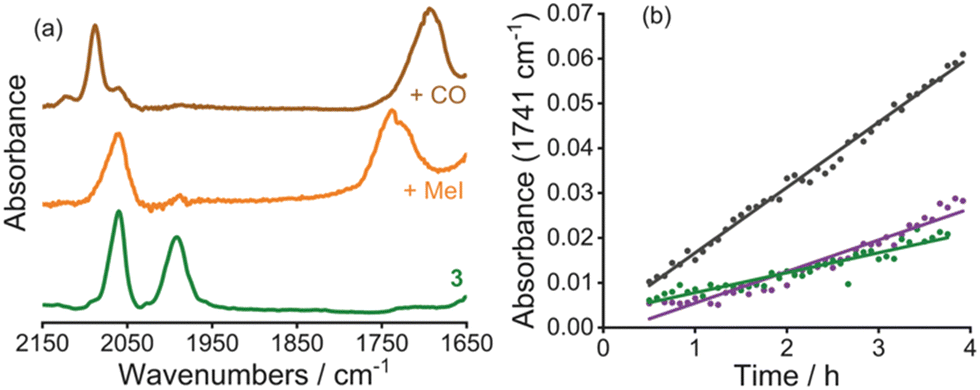 | ||
| Fig. 3 (a) Infrared spectra (ATR-IR, ν(CO) region) of 3 (green), after soaking in MeI overnight (orange), after exposure to CO atmosphere for 10 days (brown), and (b) growth of ν(CO) absorbance of methyl acetate with time using 3 (green), repeat run using solid recovered from experiment with 3 (purple) and nBu4N[Rh(CO)2I2] (grey) in CHCl3 at 120 °C (10 bar CO, 0.8 M MeI, 2.5 M MeOH). | ||
The carbonylation of MeOH/MeI catalysed by 3 was followed by in situ high-pressure IR spectroscopy, using a cylindrical internal reflectance (CIR) cell (S2.11, ESI†) as described previously.11 During the reaction (at 120 °C, 10 bar CO in CHCl3), growth of an absorption at 1741 cm−1 was observed (Fig. 3b and Fig S18, ESI†) which corresponds to the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) absorption of methyl acetate. This results from carbonylation of MeOH/MeI according to the overall reactions 2MeOH + CO → MeOAc + H2O or MeOH + MeI + CO → MeOAc + HI. The homogeneous catalytic reaction using n-Bu4N[Rh(CO)2I2] (with equimolar Rh) was also monitored under identical conditions. The TOF values determined from the slopes of the plots in Fig. 3b for the heterogeneous and homogeneous catalysts are 6.6 h−1 and 21.8 h−1, respectively. By comparison, a TOF of ∼20 h−1 was found in our recent study using a dispersible microporous polymer support.11 The slower rate for the MOF-supported catalyst may indicate greater diffusion limitations for the MOF. PXRD of the recovered material following catalysis indicates crystallinity of the MOF is maintained (Fig. 2a) and an IR spectrum (Fig. S20, ESI†) shows a ν(CO) band at 2086 cm−1 consistent with the presence of the Rh(III) acetyl complex trans-[Rh(CO)2(COMe)I3]−, as expected upon exposure of [Rh(CO)2I2]− to MeI and CO. Furthermore, a repeat experiment using the solid material recovered from the catalytic run using 3 shows no loss of catalytic activity (Fig. 3b).
O) absorption of methyl acetate. This results from carbonylation of MeOH/MeI according to the overall reactions 2MeOH + CO → MeOAc + H2O or MeOH + MeI + CO → MeOAc + HI. The homogeneous catalytic reaction using n-Bu4N[Rh(CO)2I2] (with equimolar Rh) was also monitored under identical conditions. The TOF values determined from the slopes of the plots in Fig. 3b for the heterogeneous and homogeneous catalysts are 6.6 h−1 and 21.8 h−1, respectively. By comparison, a TOF of ∼20 h−1 was found in our recent study using a dispersible microporous polymer support.11 The slower rate for the MOF-supported catalyst may indicate greater diffusion limitations for the MOF. PXRD of the recovered material following catalysis indicates crystallinity of the MOF is maintained (Fig. 2a) and an IR spectrum (Fig. S20, ESI†) shows a ν(CO) band at 2086 cm−1 consistent with the presence of the Rh(III) acetyl complex trans-[Rh(CO)2(COMe)I3]−, as expected upon exposure of [Rh(CO)2I2]− to MeI and CO. Furthermore, a repeat experiment using the solid material recovered from the catalytic run using 3 shows no loss of catalytic activity (Fig. 3b).
In summary, the carbonylation catalyst cis-[Rh(CO)2I2]− has been heterogenised in the micropores of a mixed-linker UiO-66-type MOF via Coulombic interactions. The encapsulated catalyst is active for the carbonylation of MeOH and can be recovered and recycled without loss of activity or framework decomposition. IR spectroscopic studies verify formation of the same intermediate Rh species as for the established homogeneous catalytic cycle.
We thank the University of Sheffield for funding a PhD studentship (SAI).
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Shah, Acetic acid: overview and market outloook, Indian Petrochem Conference, 2014 Search PubMed.
- A. Haynes, Advances in Catalysis, ed. B. C. Gates, H. Knozinger and F. C. Jentoft, 2010, vol. 53, pp. 1–45 Search PubMed.
- D. Förster and T. C. Singleton, J. Mol. Catal., 1982, 17, 299–314 CrossRef.
- M. J. Howard, M. D. Jones, M. S. Roberts and S. A. Taylor, Catal. Today, 1993, 18, 325–354 CrossRef.
- B. L. Smith, G. P. Torrence, M. A. Murphy and A. Aguiló, J. Mol. Catal., 1987, 39, 115–136 CrossRef CAS.
- J. H. Jones, Platinum Met. Rev., 2000, 44, 94–105 CAS.
- A. Haynes, P. M. Maitlis, G. E. Morris, G. J. Sunley, H. Adams, P. W. Badger, C. M. Bowers, D. B. Cook, P. I. P. Elliott, T. Ghaffar, H. Green, T. R. Griffin, M. Payne, J. M. Pearson, M. J. Taylor, P. W. Vickers and R. J. Watt, J. Am. Chem. Soc., 2004, 126, 2847–2861 CrossRef CAS PubMed.
- R. S. Drago, E. D. Nyberg, A. Elamma and A. Zombeck, Inorg. Chem., 1981, 20, 641–644 CrossRef CAS.
- N. Yoneda, T. Minami, J. Weiszmann and B. Spehlmann, Stud. Surf. Sci. Catal., 1999, 121, 93–98 CrossRef CAS.
- A. Haynes, P. M. Maitlis, R. Quyoum, C. Pulling, H. Adams, S. E. Spey and R. W. Strange, J. Chem. Soc., Dalton Trans., 2002, 2565–2572 RSC.
- S. A. Ivko, A. M. James, M. J. Derry, R. Dawson and A. Haynes, Catal. Sci. Technol., 2022, 12, 664–673 RSC.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS.
- V. Pascanu, G. G. Miera, A. K. Inge and B. Martin-Matute, J. Am. Chem. Soc., 2019, 141, 7223–7234 CrossRef CAS PubMed.
- M. C. Wasson, C. T. Buru, Z. Chen, T. Islamoglu and O. K. Farha, Appl. Catal., A, 2019, 586, 117214 CrossRef CAS.
- Y.-S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS PubMed.
- Z. H. Li, T. M. Rayder, L. S. Luo, J. A. Byers and C. K. Tsung, J. Am. Chem. Soc., 2018, 140, 8082–8085 CrossRef CAS.
- L. Zhang, Q. B. Cao, F. Gao, Y. P. Dong and X. F. Li, Polym. Chem., 2020, 11, 2904–2913 RSC.
- D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 CrossRef CAS PubMed.
- D. T. Genna, L. Y. Pfund, D. C. Samblanet, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, ACS Catal., 2016, 6, 3569–3574 CrossRef CAS.
- A. Grigoropoulos, A. I. McKay, A. P. Katsoulidis, R. P. Davies, A. Haynes, L. Brammer, J. Xiao, A. S. Weller and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2018, 57, 4532–4537 CrossRef CAS PubMed.
- A. Chołuj, A. Zieliński, K. Grela and M. J. Chmielewski, ACS Catal., 2016, 6, 6343–6349 CrossRef.
- A. Chołuj, R. Karczykowski and M. J. Chmielewski, Organometallics, 2019, 38, 3392–3396 CrossRef.
- A. Chołuj, P. Krzesiński, A. Ruszczyńska, E. Bulska, A. Kajetanowicz and K. Grela, Organometallics, 2019, 38, 3397–3405 CrossRef.
- Y. Liu, X. B. Xi, C. C. Ye, T. F. Gong, Z. W. Yang and Y. Cui, Angew. Chem., Int. Ed., 2014, 53, 13821–13825 CrossRef CAS PubMed.
- A. Grigoropoulos, G. F. S. Whitehead, N. Perret, A. P. Katsoulidis, F. M. Chadwick, R. P. Davies, A. Haynes, L. Brammer, A. S. Weller, J. L. Xiao and M. J. Rosseinsky, Chem. Sci., 2016, 7, 2037–2050 RSC.
- J. Y. Ren, P. C. Lan, M. Chen, W. J. Zhang and S. Q. Ma, Organometallics, 2019, 38, 3460–3465 CrossRef CAS.
- X. Wang, W. Lu, Z.-Y. Gu, Z. Wei and H.-C. Zhou, Chem. Commun., 2016, 52, 1926–1929 RSC.
- H. D. Park, M. Dincă and Y. Román-Leshkov, ACS Cent. Sci., 2017, 3, 444–448 CrossRef CAS PubMed.
- H. D. Park, M. Dincă and Y. Román-Leshkov, J. Am. Chem. Soc., 2018, 140, 10669–10672 CrossRef CAS.
- A. Burgun, C. J. Coghlan, D. M. Huang, W. Q. Chen, S. Horike, S. Kitagawa, J. F. Alvino, G. F. Metha, C. J. Sumby and C. J. Doonan, Angew. Chem., Int. Ed., 2017, 56, 8412–8416 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- M. Kandiah, S. Usseglio, S. Svelle, U. Olsbye, K. P. Lillerud and M. Tilset, J. Mater. Chem., 2010, 20, 9848–9851 RSC.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- K. Manna, T. Zhang and W. B. Lin, J. Am. Chem. Soc., 2014, 136, 6566–6569 CrossRef CAS PubMed.
- C. Wang, Z. G. Xie, K. E. deKrafft and W. L. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- J. Liang, R. P. Chen, X. Y. Wang, T. T. Liu, X. S. Wang, Y. B. Huang and R. Cao, Chem. Sci., 2017, 8, 1570–1575 RSC.
- A. Fulford, C. E. Hickey and P. M. Maitlis, J. Organomet. Chem., 1990, 398, 311–323 CrossRef CAS.
- R. J. Adcock, D. H. Nguyen, S. Ladeira, C. Le Berre, P. Serp and P. Kalck, Inorg. Chem., 2012, 51, 8670–8685 CrossRef CAS.
- A. Haynes, B. E. Mann, G. E. Morris and P. M. Maitlis, J. Am. Chem. Soc., 1993, 115, 4093–4100 CrossRef CAS.
- H. Adams, N. A. Bailey, B. E. Mann, C. P. Manuel, C. M. Spencer and A. G. Kent, J. Chem. Soc., Dalton Trans., 1988, 489–496 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of synthesis and catalysis, additional characterisation data (1H NMR; 13C{1H} CP-MAS NMR, PXRD with Pawley fits; N2 volumetric adsorption). See DOI: https://doi.org/10.1039/d2cc03087j |
| This journal is © The Royal Society of Chemistry 2022 |