
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water-in-salt electrolytes – molecular insights to the high solubility of lithium-ion salts†
Aleksandar
Tot
 and
Lars
Kloo
*
and
Lars
Kloo
*
Division of Applied Physical Chemistry, Department of Chemistry, KTH Royal Institute of Technology, Stockholm, SE-10044, Sweden. E-mail: Lakloo@kth.se
First published on 1st August 2022
Abstract
The recently established water-in-salt electrolyte (WISE) concept indicates the possible application of aqueous electrolytes in lithium-ion batteries (LiBs). The application of this type of highly concentrated electrolyte relies on a proper understanding of their thermodynamically stable solutions. Therefore, fundamental insights regarding the Li[TFSI] solubility in water are important for the rational design of reproducible and stable WISE.
Recently, increasing concerns about the safety of LiBs due to occasional fire/explosion accidents have cast a shadow on their large-scale application.1 The main cause of those safety hazards is the combination of less stable high-energy electrodes and flammable non-aqueous electrolytes in the LiBs.2,3 In that perspective, the possibility to use batteries based on water as solvent seems to be an attractive alternative. The main disadvantage for large-scale application of aqueous electrolytes in LiBs is low energy density, which is caused by the low working voltages due to electrochemical water splitting.4 A significant breakthrough in the application of aqueous electrolytes was achieved by increasing the salt concentration of salt–water mixtures, resulting in a simple and effective strategy to overcome most of the remaining hurdles limiting the present water-based LiBs.5 Since this discovery the WISE electrolytes obtained significant scientific attention followed by the concern regarding their stability.6 Very high salt concentrations result in enhanced interactions between the salt cations and anions with water, reducing the chemical activity of water molecules to undergo a reduction to unwanted hydrogen gas. Despite increase in stability, the main drawbacks of WISE involve costs, toxicity and viscosity of the electrolyte, as well as a reduction of specific energy and faster self-discharge.7–9 The commercially used Li[PF6] salt has been shown not to be suitable for aqueous electrolytes, since Li[PF6] undergoes fast hydrolysis followed by the evolution of HF, which reduces the stability window and represents a significant environmental hazard.10,11 On the other hand, lithium bis(trifluoromethane sulfonyl)imide, Li[TFSI] displays better stability in aqueous electrolytes, and the favorable hydrolytic reaction is moved towards evolution of LiF. For this reason, Li[TFSI] has become the most commonly used salt in the WISEs.12,13 This type of WISE was firstly proposed by Suo and coworkers, and since then it has become extensively used in the investigation of aqueous LiBs with reported concentrations up to 21 mol of Li[TFSI] per 1 kg of water.5 However, in the literature, there is a lack of systematic solubility data of aqueous Li[TFSI] at different temperatures. The work of Gilbert and co-workers provides the salt solubility at ambient temperature based on the simple flask-shake method, used without precise temperature control.14 The work of Ding et al. provide a full phase diagram of the Li[TFSI] + H2O system using a melting point technique showing significantly higher solubility as compared to the flask-shake method.15 The reason for discrepancies could be found in the formation of meta-stable lithium-salt hydrates. This problem is well known for lithium halides characterized by inconsistency of experimental results depending on the method of measurement used.16,17 Therefore, the alarming possibility to accidentally make an over-saturated Li[TFSI] solution by heating to saturation with subsequent cooling is certainly a risk. The potential over-saturation can cause significant issues in the reliable evaluation of battery performance when using WISE, especially regarding the long-term performance. In this work, the solubility of aqueous Li[TFSI] is presented, and the results are rationalized on the basis of spectroscopic and molecular dynamics (MD) investigations of aqueous Li[TFSI] systems at different mole ratios. In this context, it should be noted that the Li+ ion is one of the most polarizing cations, perhaps the one most similar to H+, and strong hydration with local structures not too different from those in acidic water solutions, are expected and also identified.
The solubility of Li[TFSI] in water was determined in the temperature range 288–323 K, and the results are presented in Table 1. In order to confirm that equilibrium was obtained during the experiments, the samples were extracted at different equilibration times.
| Temperature (K) | Molality (mol of Li[TFSI] per kg of water) | Li[TFSI]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O molar ratio :H2O molar ratio |
|---|---|---|
| 288 | 14.23 (±0.13) | 1:3.90 |
| 293 | 16.24 (±0.22) | 1:3.42 |
| 298 | 18.41 (±0.28) | 1:2.97 |
| 303 | 20.05 (±0.11) | 1:2.77 |
| 308 | 22.43 (±0.25) | 1:2.48 |
| 313 | 25.06 (±0.36) | 1:2.22 |
| 318 | 26.84 (±0.23) | 1:2.10 |
| 323 | 27.21 (±0.18) | 1:2.04 |
The solubility was determined by gravimetry, while the co-existing solid phase was identified using X-ray power diffraction (XRPD). Equilibrium is postulated when the changes in the solubility and XRPD traces no longer can be observed. A typical diffractogram is presented in Fig. S1 (ESI†). The XRPD traces of the solid phase observed as a function of the scattering angle 2θ show prominent peaks at 14.25°, 16.01°, 18.43°, 19.01°, and 21.5°, which all are in good agreement with the literature values regarding pure Li[TFSI](s).18 The obtained scattering traces were used to confirm that an equilibrium between dissolved Li[TFSI] and solid Li[TFSI] was obtained at each temperature investigated displaying a solubility range from 14.23 mol kg−1 at the lowest temperature to 27.21 mol kg−1 at the highest temperature. The solubility at 293 K (16.24 mol kg−1) is slightly higher as compared to that reported by Gilbert et al., 16.01 mol kg−1 (or 0.8217 wt%) at, as stated, 295 K.11 In their work, the authors also applied the shake-flask method, but without precise temperature control and with no specification of the time of equilibration. Furthermore, the solubility obtained at 298 K in this work (18.41 mol kg−1) is significantly lower in comparison to the results of Suo et al., where it was suggested that a molality higher than 20 mol kg−1 of aqueous Li[TFSI] can be obtained at the stated temperature.5 The origin of this discrepancy cannot be determined, since the details of their solubility determination are not provided. Nevertheless, these acclaimed high solubilites should be taken with some reservation.
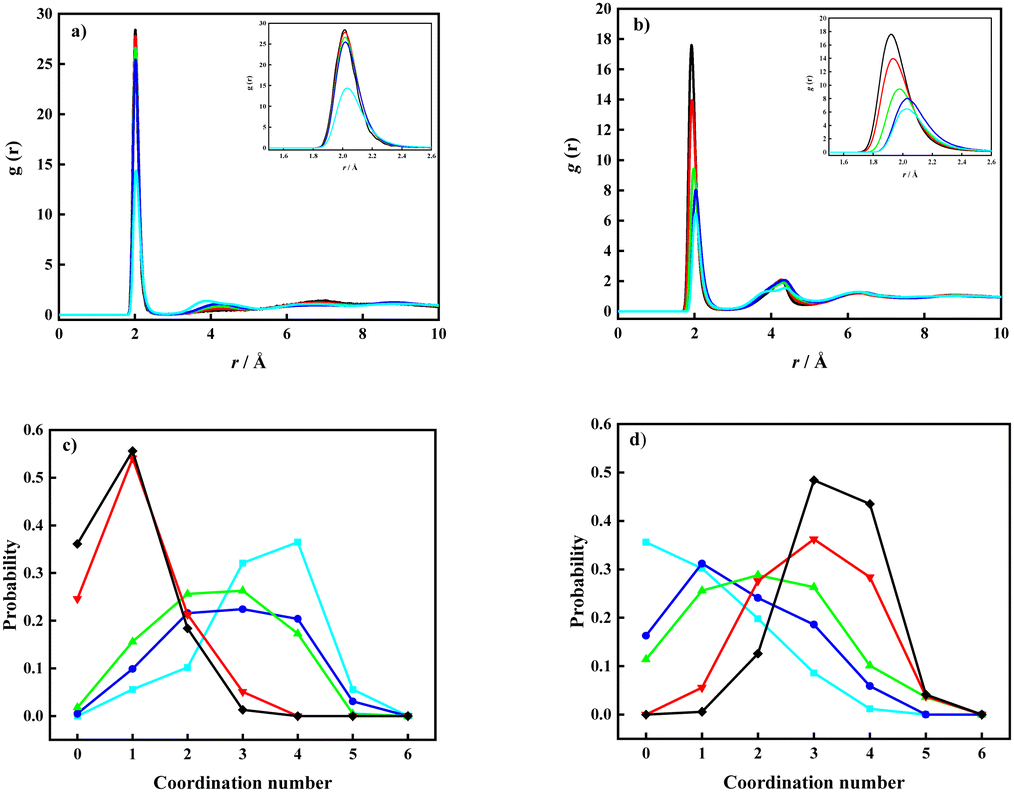
Experimentally determined solubilities have a story to tell, and general solution chemistry knowledge suggests that the hydration of, in particular, the lithium cations must play a central role. MD simulations can be used as a helpful tool to visualize and to determine the dependence of salt solubility with respect to the solvation environment of the salt ions. Therefore, a systematic study at different ratios of Li-ion salt and water was performed, based both on the systems that were shown to be soluble in the water (molar ratios of 1:3, 1:4, and 1:6, for the Li[TFSI]:H2O mixtures), as well as for hypothetical systems that exceed the limiting solubility (molar ratios 1:2 and 1:1). The number of ions and water molecules included in the models in each simulation are presented in Table S1 (ESI†). The solvation environments of water and Li+ were studied in more detail based on the MD simulation results by estimates of partial radial distribution functions (pRDFs) and coordination numbers (CNs). CNs represents the average coordination number during the simulation and were estimated using the equation:
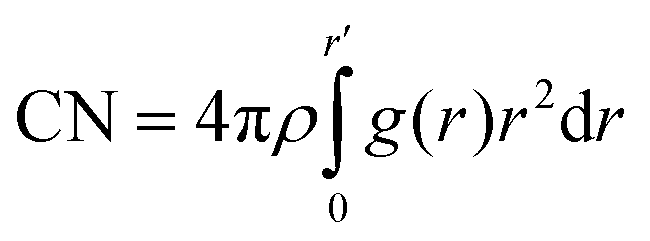 | (1) |
First, a water organization could be observed in the water O–O pRDFs and CNs. The obtained results are presented in Fig. 1 and Fig. S2 (ESI†). The height of the first peak in the pRDFs shows a decrease with the increase of salt concentration, followed by a shift of the maximum towards a longer distance. The shortest distance between oxygen atoms in the water molecules was determined to 2.74 Å for Li[TFSI](1):H2O(6), with a significant increase of the distance to 2.90 Å (Li[TFSI](1):H2O(4)), increasing further to 3.06 (Li[TFSI](1):H2O(3)), as the molar fraction of salt was increased. A further decrease of the amount of water offered a less pronounced effect in pRDF peak shifts (3.10 Å and 3.12 Å). Also, it can be noted that the second peak area, residing at a distance of ∼5.5 Å, decreases as the water content is decreased. Notable peaks can be observed for liquid systems (Li[TFSI](1):H2O(6), Li[TFSI](1):H2O(4), and Li[TFSI](1):H2O(3)), while this second peak is missing all together in the partial RDFs of the Li[TFSI](1):H2O(2) and Li[TFSI](1):H2O(1) mixtures. As a conclusion, the most significant difference with respect to the O–O partial RDF peaks can be observed between systems below and above the experimentally determined solubility limit. The two pRDF peaks can be assigned to originate from the tetrahedral structure of assemblies of water molecules in the mixtures, where each water molecule is surrounded by four water molecules like in pure water. The peaks that correspond to the distance of ∼2.8 Å originate from coordination of the “central” water molecule by neighbouring water molecules, while the peaks at a distance ∼5.5 Å represent O–O distances between the next-nearest neighbour water molecules coordinated to the central water molecule. The literature suggests that the addition of Li[TFSI] causes a substitution of the central water molecule by Li+ ions, causing the observed change in the next-neighbour water–water molecule distance (decrease in the second-peak area).19 The water network structure was also monitored by the calculation of an O–O coordination number (Fig. S2, ESI†). The water–water CN continuously decreases with increasing Li[TFSI] concentration, as shown in Fig. S2 (ESI†). The probability of the O–O CNs represents a more dynamic representation of the solvation environment, and it was determined for each system and the values are presented in Fig. 1b.
 | ||
| Fig. 1 Partial RDFs (a), and coordination number probability (b) of water O–O atoms. Black – Li[TFSI](1):H2O(1); red – Li[TFSI](1):H2O(2); green – Li[TFSI](1):H2O(3); blue – Li[TFSI](1):H2O(4); cyan – Li[TFSI](1):H2O(6). | ||
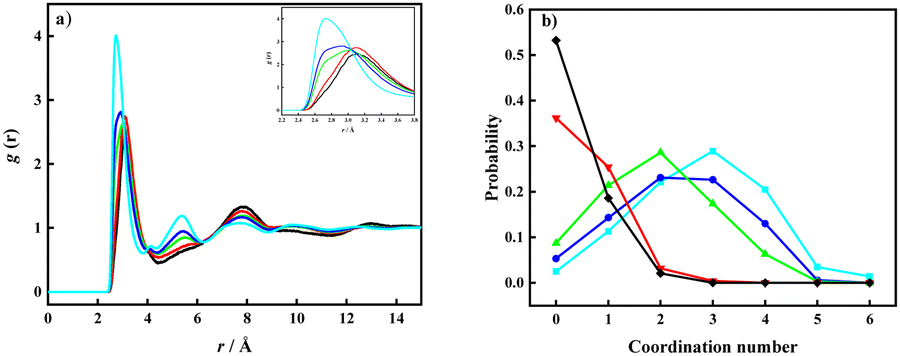
The probability distribution of the CNs was calculated from each time step in the simulations with a cut-off distance <4.0 Å. The addition of Li[TFSI] leads to a less pronounced organization of water molecules, followed by a shift of CN towards lower probabilities. Nevertheless, the obtained results suggest that systems with a concentration of lithium-ion salt below the solubility limit display a wide distribution in water–water CNs while above the solubility limit the CNs collapse to low values. This suggests that precipitation of Li[TFSI] takes place when the amount of water molecules becomes insufficient to maintain a local water structure. In parallel, also the Li-ion solvation was investigated. The pRDFs monitoring the Li+ solvation by O(TFSI) and O(water) are shown in Fig. 2a and b. By comparing the pRDFs for Li–water and Li–TFSI interactions, both systems give rise to a sharp peak at about the same distance (≈2 Å). The intensity of the peaks increases gradually with the concentration of Li[TFSI] with respect to O(TFSI)–Li+, while the O(water)–Li+ intensity significantly increases going from Li[TFSI](1):H2O(6) to Li[TFSI](1):H2O(4). Further increase of Li-ion salt concentration has insignificant influence on the intensity of the short-distance peak. In addition, it can be observed that the first peak clearly shifts with respect to the O(TFSI)–Li+ contact, while the distance of the O(water)–Li+ interaction remains constant. Following the CN probability (Fig. 2c and d) estimated with a cut-off distance of 3.0 Å from the Li+ ion, a significant change regarding water molecules coordinated to the lithium ions is observed only after exceeding the experimentally determined solubility limit. Below the solubility limit, Li+ shows a broad range of coordination numbers (from 1 to 3) with about the same probability. Similar results were obtained for the overall coordination numbers in the work of Zhang et al.19 and Borodin et al.20 In the systems Li[TFSI](1):H2O(1) and Li[TFSI](1):H2O(2) instead a significant decrease in the CN of water–Li+ coordination can be observed, where the highest probability can be assigned to a system with only one water molecule in the lithium-ion solvation shell. In addition, in these two super-concentrated mixtures, the probability to find lithium at all without any water in the coordination sphere becomes high; amounting to almost 25% (Li[TFSI](1):H2O(2)) and 35% (Li[TFSI](1):H2O(1)), while in the other systems corresponding to concentrations below the solubility limit this probability is close to 0%. With an increasing salt concentration in the solution, Li+ and TFSI− start to form more frequent direct contacts, as indicated by the increase in the CN for O(TFSI)–Li+. At the highest concentration of Li[TFSI] corresponding to a homogenous mixture (Li[TFSI](1):H2O(3)), close to 90% of the Li+ ions are coordinated by TFSI− corresponding to a broad range of CNs. At higher salt concentrations, essentially all lithium ions are coordinated by TFSI−. Therefore, once salt concentrations become so high that the probability to find less than 2 water molecules in the solvation shell of lithium ions becomes high, precipitation spontaneously takes place.
 | ||
| Fig. 2 Partial RDFs of O(water)–Li+ (a), O(TFSI)–Li+ (b), and coordination number probability for O(water)–Li+ (c) and O(TFSI)–Li+ (d). Black – Li[TFSI](1):H2O(1); red – Li[TFSI](1):H2O(2); green – Li[TFSI](1):H2O(3); blue – Li[TFSI](1):H2O(4); cyan – Li[TFSI](1):H2O(6). | ||
The exclusion of water molecules from the Li-ion solvation sphere can also be followed in Raman spectra of the mixtures, where a significant shift in the Raman bands of the S–N–S bending modes (745 cm−1) of the TFSI anions (Fig. S3, ESI†) can be noted.5 It has been shown that this Raman peak from the anion is highly suitable to monitor the Li-ion–TFSI interaction, and it shifts towards higher wavenumbers as the salt concentration is increased. Wavenumbers higher than 749 cm−1 have in the literature been ascribed to aggregate ion-pairs of lithium cations and TFSI− anions, which in this work is observed for the oversaturated systems. Moreover, the changes of the O–H stretching vibration (2800–3500 cm−1) of water upon addition of Li[TFSI] was investigated by using the principle of excess Raman spectra.21 The excess spectra were obtained by subtracting the pure water Raman spectra from the spectra of lithium–salt containing solutions. In the excess spectra positive and negative bands can be observed (Fig. S4, ESI†). The positive band in excess spectra represents the water molecule that interacts with the added ions, while the negative band is suggested ta mainly arise from the depletion of bulk water. The (negative) integral area of the negative peak is gradually increased by the addition of Li[TFSI] suggesting the progressive disturbance of the bulk water structure. The hydration number (Nhydration) can be estimated from the hydration spectra (Fig. S5, ESI†), based on the equation:
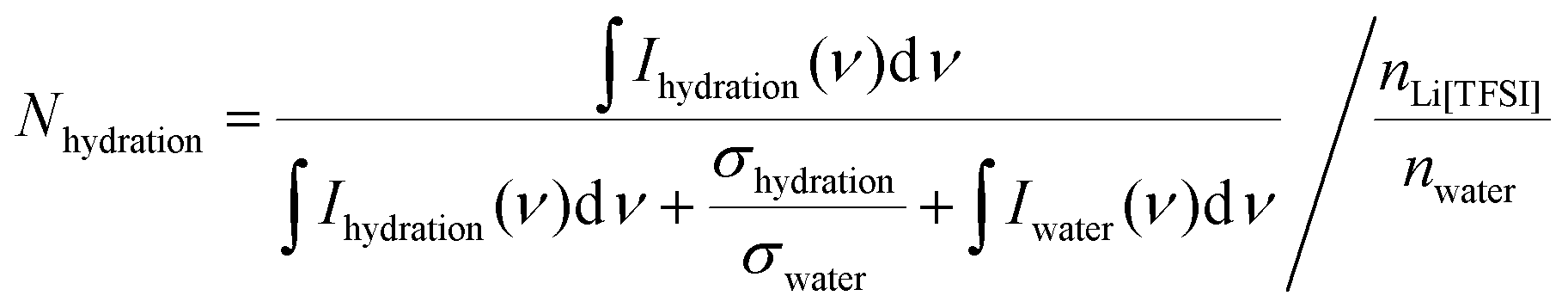 | (2) |
 | ||
| Fig. 3 The hydration number calculated from Raman spectra. | ||
The results show that an insufficient amount of water molecules with respect to the solvation of the lithium cation causes the experimentally observed precipitation and results from the MD simulations offer insights to the reasons at a molecular level. The fundamental results for the Li[TFSI] solubility in water presented in this work are important for the rational design of reproducible and stable WISEs. The obtained solubility of 18.41 mol kg−1 at 298 K suggests that the Li[TFSI] concentration in WISEs should be carefully selected. The use of thermodynamically equilibrated solutions allows better control of the electrochemical processes and will significantly help in the understanding of the processes that increase the stability of aqueous electrolytes. The MD simulations (Fig. 4) suggest that the mole ratio of Li[TFSI] to water corresponding to 1:3 forms an upper limit for stable mixtures, in which a sufficient amount of water are available for, at least partially, solvate the Li+ cations.
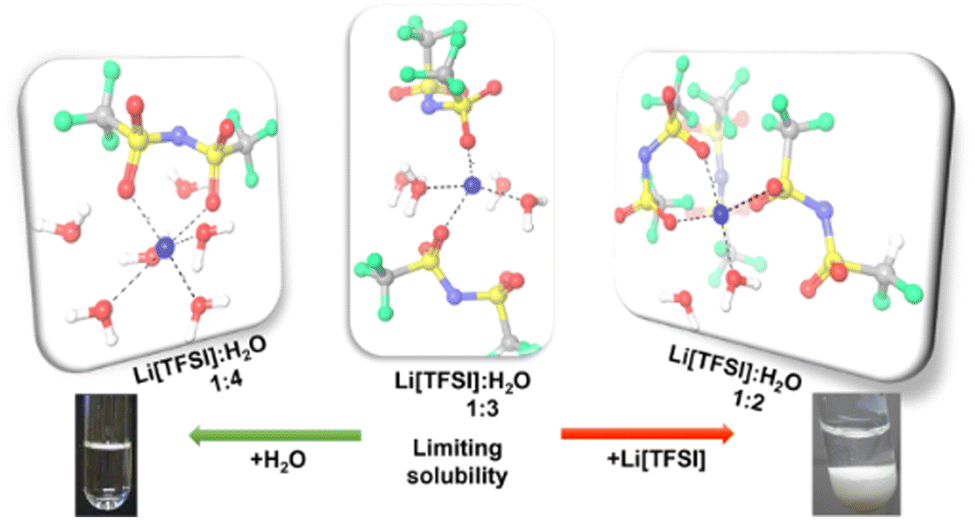 | ||
| Fig. 4 The representative snapshots from MD simulations. | ||
The work was supported by the Swedish Energy Agency contract no. 50119-1, entitled “Be WiSE” and the Swedish Research Council contract no. 2020-06701.
Conflicts of interest
There are no conflicts to declare.References
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484 RSC.
- K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef CAS PubMed.
- H. Kim, J. Hong, K. Y. Park, H. Kim, S. W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788 CrossRef CAS PubMed.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938 CrossRef CAS PubMed.
- L. Droguet, A. Grimaud, O. Fontaine and J. M. Tarascon, Adv. Energy Mater., 2020, 10, 2002440 CrossRef CAS.
- D. Chao and S. Z. Qiao, Joule, 2020, 4, 1846–1851 CrossRef.
- J. Xu and C. Wang, J. Electrochem. Soc., 2022, 169, 030530 CrossRef.
- D. Chao, W. Zhao, F. Xie, C. Ye, H. Li, M. Jaroniec and S. Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed.
- A. Hammami, N. Raymond and M. Armand, Nature, 2003, 424, 635 CrossRef CAS PubMed.
- Y. Ha, C. Stetson, S. P. Harvey, G. Teeter, B. J. Tremolet de Villers, C. S. Jiang, M. Schnabel, P. Stradins, A. Burrell and S. D. Han, ACS Appl. Mater. Interfaces, 2020, 12, 49563 CrossRef CAS PubMed.
- S. F. Lux, L. Terborg, O. Hachmöller, T. Placke, H. W. Meyer, S. Passerini, M. Winted and S. Nowak, J. Electrochem. Soc., 2013, 160, A1694 CrossRef CAS.
- L. Suo, Y. S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- W. J. R. Gilbert, J. Safarov, D. L. Minnick, M. A. Rocha, E. P. Hassel and M. B. Shiflett, J. Chem. Eng. Data, 2017, 62, 2056 CrossRef CAS.
- M. S. Ding and K. Xu, J. Phys. Chem. C, 2018, 122, 16624–16629 CrossRef CAS.
- J. P. Simmons, H. Freimuth and H. Russell, J. Am. Chem. Soc., 1936, 58, 1692–1695 CrossRef CAS.
- D. A. Boryta, J. Chem. Eng. Data, 1970, 15, 142–144 CrossRef CAS.
- J. Bao, X. Qu, G. Qi, Q. Huang, S. Wu, C. Tao, M. Gao and C. Chen, Solid State Ionics, 2018, 320, 55–63 CrossRef CAS.
- Y. Zhang, N. H. C. Lewis, J. Mars, G. Wang, N. J. Weadock, C. J. Takacs, M. R. Lukatskaya, H. G. Steinrück, M. F. Toney, A. Tokmakoff and E. J. Maginn, J. Phys. Chem. B, 2021, 125, 4501 CrossRef CAS PubMed.
- O. Borodin, L. Suo, M. Gobet, X. Ren, F. Wang, A. Faraone, J. Peng, M. Olguin, M. Schroeder, M. S. Ding, E. Gobrogge, A. von Wald Cresce, S. Munzos, J. A. Dura, S. Greenbaum, C. Wang and K. Xu, ACS Nano, 2017, 11, 10462–10471 CrossRef CAS PubMed.
- H. Ge, Y. Zhao, H. Yang and M. Wang, Spectrochim. Acta, Part A, 2022, 267, 120543 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc03062d |
| This journal is © The Royal Society of Chemistry 2022 |