
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A pH-responsive crosslinker platform for antibody-drug conjugate (ADC) targeting delivery†
Francesca
Migliorini
,
Elena
Cini
,
Elena
Dreassi
,
Federica
Finetti
,
Giovanni
Ievoli
,
Giulia
Macrì
,
Elena
Petricci
 ,
Enrico
Rango
,
Lorenza
Trabalzini
and
Maurizio
Taddei
*
,
Enrico
Rango
,
Lorenza
Trabalzini
and
Maurizio
Taddei
*
Dipartimento di Biotecnologie, Chimica e Farmacia, Università degli Studi di Siena, Via A.Moro 2, 53100 Siena, Italy. E-mail: maurizio.taddei@unisi.it
First published on 31st August 2022
Abstract
We report a new 1–6 self-immolative, traceless crosslinker derived from the natural product gallic acid. The linker acts through a pH-dependent mechanism for drug release. This 5-(hydroxymethyl)pyrogallol orthoester derivative (HMPO) was stable for 24 hours at pH values of 7.4 and 6.6 and in plasma, releasing molecules bound to the hydroxymethyl moiety under acid-dependent stimuli at pH 5.5. The linker was non-toxic and was used for the conjugation of Doxorubicin (Doxo) or Combretastatin A4 with Cetuximab. The ADCs formed showed their pH responsivity reducing cell viability of A431 and A549 cancer cells better than Cetuximab alone.
Conjugation between a targeting moiety and a drug is one of the most efficient methods for overcoming drug selectivity problems.1 Besides antibody-drug conjugates (ADCs),2 there are excellent examples of conjugates containing integrin,3 carbonic anhydrase ligands,4 folic acid,5 prostate specific antigen (PSA)6 or somatostatin.7 Conjugation allows an increase in drug concentration in the tissues where the target is located after the drug cargo has been selectively activated at the site of action. Release is controlled by a trigger mechanism that depends on the linker, the key component of the conjugation system.8 The way the linker is removed (and the drug released) depends on the trigger mechanism. Historically, the first external stimulus used to release drugs from a ADC was pH.9 In cancer, mechanisms related to abnormal rapid metabolism and proliferation lead to a hypoxic microenvironment in which several acidic molecules are produced that lower the pH to around 5.7–6.9.10 Within the tumour cell, the proton influx lowers the intracellular and substructural pH to 5.0–6.0, while a lower pH (4.0–5.0) is achieved in the lysosomes.11 The pH difference between tumour cells and healthy tissue has been exploited to produce pH-responsive anti-cancer nanomaterials,12 although the presence of an acidic tumour microenvironment poses a potential problem for selectivity. At ADC, once the antibody reaches its target, the internalisation of the receptor selectively transports the conjugate into the interior of the cell to be eventually metabolised in the acid lysosome environment.2a FDA-approved acylhydrazones (found in Mylotarg® and Besponsa®), for example, release the active ingredient in acidic environments, but can only be made from carbonyl or hydrazine derivatives, limiting the delivery to agents that contain these functions.13 The bifunctional cross-linker N-ethoxybenzylimidazole (NEBI) has been used as a tunable pH-sensitive linker and used for targeted delivery of indenoisoquinoline agents or modified Doxo to cancer tissue (Scheme 1).5d,14 In this case, a benzaldehyde or an imidazole moiety remains in the released drug. Maleimido derivatives, after hydrolytic conversion to monoamides of maleic acid, have a proximal carboxylate group that supports amide hydrolysis with the formation of the malic anhydride under acidic conditions.15 Although efficient, this linker is limited to transporting only primary amines (Scheme 1).1
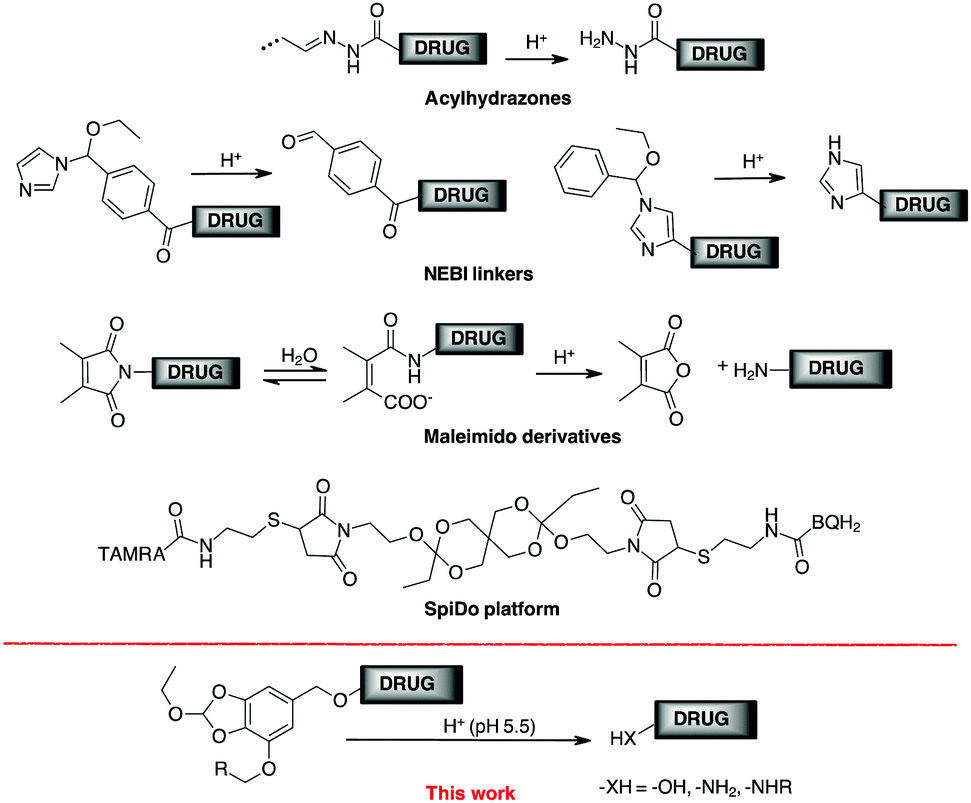 | ||
| Scheme 1 Examples of acid labile linkers for drug targeting. | ||
Wagner and co-workers have determined the release kinetics of several functional groups between pH 7.4 and 5.5 and classified their properties in terms of behaviour under neutral conditions.16 The spiro-orthoester SpiDo exhibited a hydrolysis half-life t1/2 = 1.5 h (assuming a first-order reaction) and complete hydrolysis was obtained after 7 h at pH 5.5.17 However, to our knowledge, the SpiDo platform has only been used for in vitro imaging experiments with microorganelles.
In conclusion, despite the great improvements achieved in pH-sensitive linkers, a new chemistry for versatile linkers with adjustable release rate is still highly desirable for prodrug development, including ADC and gene delivery.
The 1–6 self-immolative elimination process, typical of p-amino or p-hydroxybenzyl alcohol, is one of the most common processes to drive the release of prodrugs and bioconjugates by external stimuli.18 We report here a new linker based on the 1–6 self-immolative p-hydroxybenzyl alcohol platform designed for the conjugation of a drug to a target system (Scheme 1) and the corresponding release at acid pH. The orthoester group was chosen to provide an effective trigger at pH around 5.5, and the benzyl alcohol moiety was used for binding drugs bearing an amine (as a carbamate) or a phenol (as an aryl benzyl ether). In one side of the aromatic ring, we incorporated a triple bond for click chemistry to bind the cargo to the antibody support. The value of this platform was evaluated in the analysis of cytotoxic drug delivery to cancer cells by conjugation with cetuximab (Ctx), a monoclonal antibody (mAb) specific for epidermal growth factor receptor (EGFR).
Since orthoesters are known to be stable in plasma but sensitive to hydrolysis at pH 5.5,16,19 we developed an aromatic orthoester derived from gallic acid, a natural tannin component found in several plants and available in large quantities at a low price. Gallic acid (1, Scheme 2) already has all the properties for our design: two OH groups to form a cyclic orthoester, another OH for the installation of a triple bond for orthogonal couplings, and the p-carboxyl group that can be easily reduced to benzyl alcohol. Thus, gallic acid 1 was esterified to the corresponding ethyl ester 2, which cyclised with triethyl orthoformate to give the orthoester 3 in high yield (Scheme 2). The remaining free –OH reacted with propargyl bromide 4 (K2CO3/KI in dry acetone for 2 h) and the propargyl ether was reduced with LiAlH4 in THF to give the 5-(hydroxymethyl)pyrogallol orthoester derivative 5 (HMPO) in good overall yield starting from 1 (Scheme 2).
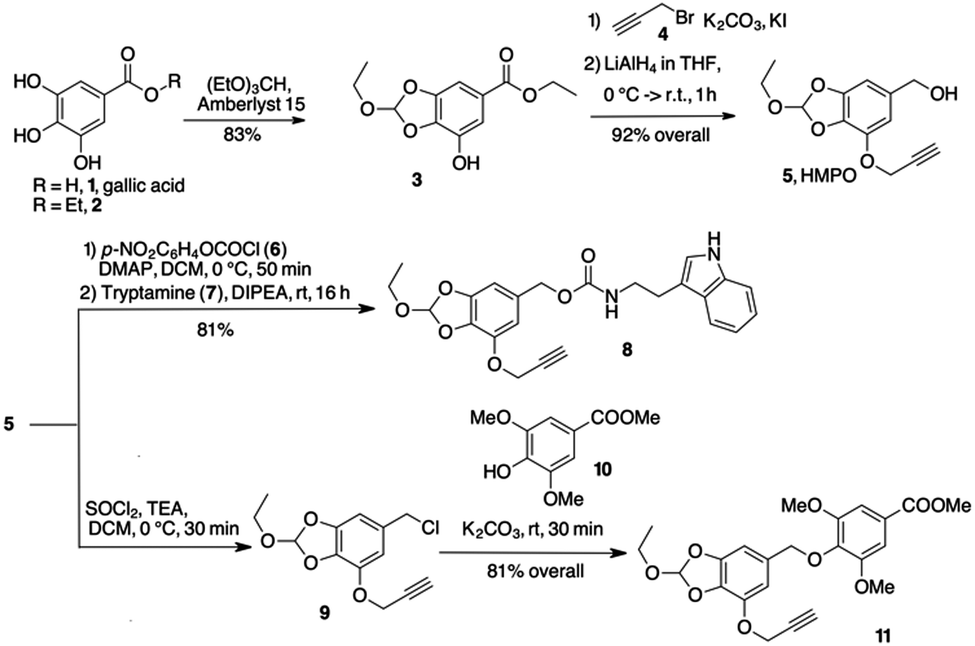 | ||
| Scheme 2 Synthesis and linkage of HMPO platform. | ||
Compounds 8 and 11 (Scheme 2) were prepared to evaluate the hydrolysis profile and release of the molecule linked to the benzyloxy group. Activation of 5 with p-nitrophenyl-chloroformate and further reaction with tryptamine 7 in alkaline medium gave carbamate 8 in acceptable yields. After conversion of alcohol 5 to benzyl chloride 9 (see ESI†), nucleophilic displacement with 10 gave phenyl ether 11. The behaviour of compounds 8 and 11 at different pH values was investigated by HPLC analysis. Compound 8 released the payload almost completely after 7 hours at pH 5.5 (95% release, t(1/2) = 3 h, Fig. S1, ESI†). A comparable kinetics was observed at the same pH with 11 (t1/2 ≅ 3 h and the 95% of release reached after 6 h, Fig. S2, ESI†). When 8 and 11 were treated at 37 °C at pH 7.4 and 6.5, no significant hydrolysis occurred within 6 h.
Only after 24 h at pH 6.5 the HPLC profile of the samples showed the presence of 10% of tryptamine 7 and 25% of phenol 10 respectively (Fig. S1 and S2, ESI†). These results agreed with those reported for existing orthoester linkers, confirming that HMPO is sensitive to lysosomal pH, showing an excellent stability profile at physiological and extracellular solid tumor pH values.
To get a plausible picture of the disassembling mechanism for HMPO conjugates, we studied the behavior of 11 at pH 5.5 through 1H NMR analysis. The sample was incubated in deuterated acetic buffer at 37 °C and the FID recorded every 30 minutes. After 5 minutes, the shifts of the aromatic signals from δ 7.87 to 7.92 and of the methoxy singlet from δ 4.42 to 4.49 suggested the release of phenol 10 in solution (Fig. S3, ESI†). Moreover, we noticed a new peak at δ 8.69 assigned to the transient assembling of a formate derivative. Finally, the characteristic aromatic and aliphatic signals of a 5-(hydroxymethyl)-3-(2-propynyloxy)pyrogallol (12, Scheme 3) were observed, confirming the complete disassembling of the structure and the release of the phenol. An ESI/MS analysis of the solution contained in the NMR tube confirmed the presence of compounds 10 and 12.
 | ||
| Scheme 3 Proposed mechanism for disassembling of HMPO cargo. | ||
A reasonable mechanism for cargo release from the HMPO platform was then supposed (Scheme 3). In acid conditions, the orthoester moiety is protonated releasing EtOH. The transient (stabilized) carbocation 11b gives the pyrogallol formate 11f. The expected 1–6 elimination occurs with formation of p-quinone methide 11g and release of the cargo 10.
The alternative cleavage of the protonated 2-hydroxy-benzodioxole 11d might give formate 11h that, after ester hydrolysis, releases the cargo 10 through 1–6 elimination.
We applied the HMPO platform to the conjugation of the antitumor drugs Doxo (13) and Combretastatin A4 (14) with Cetuximab (Ctx), a monoclonal antibody specific for the epidermal growth factor receptor (EGFR). Activation of 5 with p-nitrophenyl chloroformate and further nucleophilic displacement with Doxo gave carbamate 15 in good yield (73%) (Scheme 4). Compound 15 was subjected to CuCAAC reaction with 6-azidohexanoic acid 16 in the presence of Cu(II) acetate and sodium ascorbate in DMF/water, providing compound 17 in 64% yield (Scheme 4). Alternatively, chloride 9 reacted with Combretastatin A4 (14), deprotonated by NaH in DMF, to give compound 18 in a good yield (98%). Afterwards, azide 16 gave 19 in 75% yield (Scheme 4).
 | ||
| Scheme 4 Conjugation of Doxo and Combretastatin A4 with HMPO. | ||
Molecules 15 and 18 resulted stable in buffers at pH 7.4 and 6.5 (Table S1 and Fig. S4, S5, ESI†). Only a small amount of free Doxo (4%) and Combretastaine A4 (5%) was detected after 24 h at 37 °C at pH 7.4.
The stability in water was investigated to exclude false positive release, confirming that 15 and 18 remain untouched after 24 h at 37 °C in water. A test was also done in human plasma showing a good stability of these compounds (Table 1). The payload 15 showed a t1/2 value in line with the general behavior of pure Doxo in plasma.20 These results confirmed that pH-sensitive HMPO platform is stable to biological fluids as blood. On the other hand, hydrolysis at pH 5.5 showed a rapid release of drugs 13 and 14 (Fig. S4 and S5, ESI†). With 15, in the first hours a plateau was reached and almost 90% of drug was released after 6 h. Although with a different behaviour, 93% of Combretastatin A4 (14) was released from 18 after 12 h (see ESI†).
| Comp. | H2Oa | pH 7.4,at1/2b | pH 6.5,at1/2b | Plasma,at1/2b |
|---|---|---|---|---|
| a Value expressed as percentage of unmodified compound after 24 h of incubation. b Half-life (t1/2) expressed as the amount of time it takes before half of the drug is hydrolyzed/degraded. | ||||
| 15 | 99% | 94%, >24 h | 90%, >24 h | 35%, 8.3 h |
| 18 | 99% | 95%, >24 h | 93%, >24 h | 89%, 27.6 h |
For the preparation of the ADCs, carboxylic acids 17 and 19 were transformed into the corresponding NHS-esters in PBS at pH 7.4 in presence of Sulfo-NHS (20) and EDC, and further reacted with Ctx (Scheme 5) to obtain bioconjugates 21 and 22. After purification by dialysis, the drug antibody ratio (DAR) was determined by MALDI analysis (Table S1, ESI†). A HPLC/MS analysis of the dialyzed solution showed the absence of the free drugs 13 and 14 in solution. The anti-proliferative activity (MTT assay) of the conjugate 21 and 22 was evaluated in A549 (human lung carcinoma, Fig. 1) and A431 (epidermoid carcinoma cell line, Fig. S7, ESI†), both overexpressing the epidermal growth factor receptor (EGFR). Treatment with the two conjugates showed a stronger antiproliferative effect if compared with unconjugated Ctx (Fig. 1). The effect was also comparable to the activity of the free drugs confirming the release of Doxo and Combretastatin A4 from the linker in the cell culture. The observed differences between the release kinetics of 15 and 18 and the cell viability assays of the corresponding conjugates 21 and 22 might be due to the different mechanism of action of the two drugs (intercalation into DNA and inhibition of topoisomerase II for Doxo versus depolymerisation of tubulin for Combretastatin A4), which probably requires different times of action.
 | ||
| Scheme 5 ADC preparation. | ||
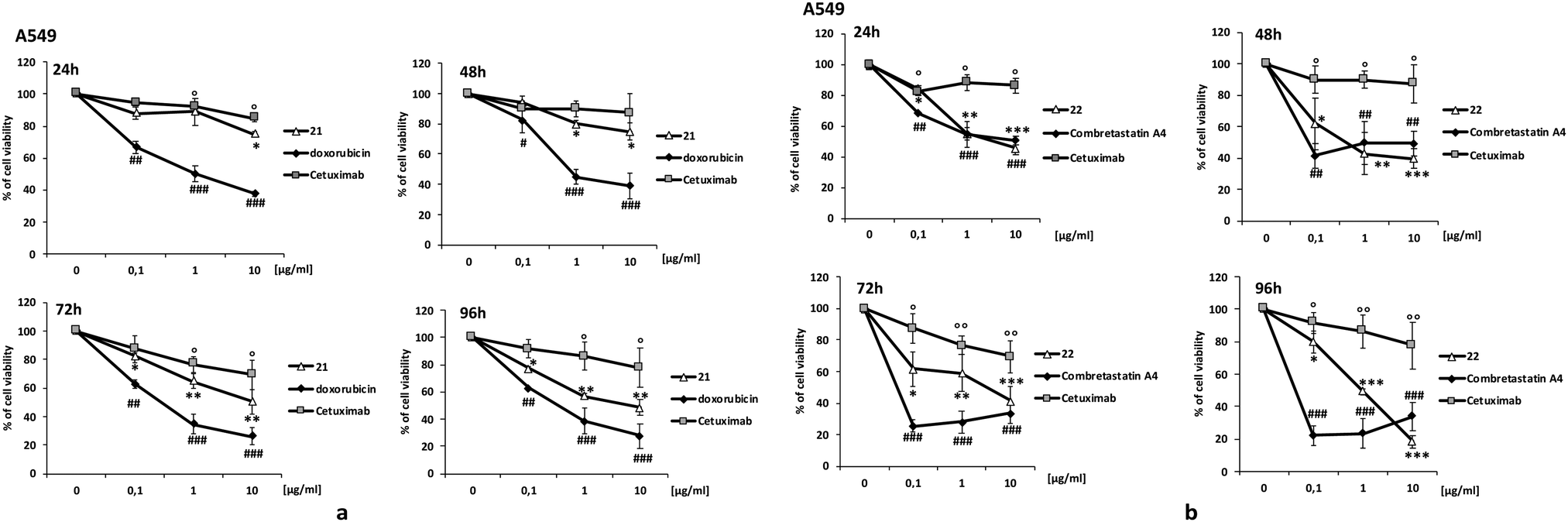 | ||
| Fig. 1 Cell viability. Cancer cell survival was evaluated by MTT test. Cells were exposed to increasing concentration of: (a) conjugate 21, Doxorubicin and Cetuximab; (b) conjugate 22, Combretastatin A4 and Cetuximab. Data are expressed as percent of cell viability. #,*, ° p < 0.05 vs. basal; ##,**, °° p < 0.01 vs. basal and ###,***, °°° p < 0.001 vs. basal. | ||
HMPO alone wasn’t toxic, on the other hand, on cells employed in the tests. DAPI staining, Annexin V-FITC and PI staining tests were also employed to investigate the pathway of cell death. The results showed that 21 and 22 significantly induce apoptosis and cell death (Fig. S7 and S8, ESI†).
In conclusion, we demonstrated that the new HMPO platform (5) can be employed for drug delivery in acid biologic environments resulting suitable for conjugation of the cytotoxic drugs Doxo (a primary amine) and Combretastatin A4 (a phenol) with an antibody carrier. The resulting ADCs exhibited improved cytotoxic activity in cancer cells compared to the unconjugated antibody and to the cargo-linker fragment.
Various primary and secondary amine-, alcohol- and phenol-containing molecules can be bound by the formation of carbamates, carbonates or ethers. Orthogonal reactivity to these bonds is possible by introducing azides, alkynes, tetrazines or other fragments for click chemistry to allow easy conjugation with macromolecular supports. HMPO has the same stability and other typical properties as other pH-reactive linkers, but is non-toxic and easily accessible synthetically (4 steps, 76% overall yield) from gallic acid, a cheap natural starting material. Ultimately, the low toxicity suggests a potential use of our linker in non-oncology therapies. Application of the linker to non-canonical cytotoxic drugs for intra- and extracellular delivery is in preparation and will be reported shortly.
This work was supported by the AIRC (under IG 2017 – ID. 20758 project – P. I. Taddei Maurizio).
Conflicts of interest
There are no conflicts to declare.Notes and references
- X. Yang, Z. Pan, M. R. Choudhury, Z. Yuan, A. Anifowose, B. Yu, W. Wang and B. Wang, Med. Res. Rev., 2020, 40, 2682–2713 CrossRef PubMed.
- (a) K. C. Nicolaou and S. Rigol, Angew. Chem., Int. Ed., 2019, 58, 11206–11241 CrossRef CAS PubMed; (b) K. Tsuchikama and Z. An, Protein Cell, 2018, 9, 33–46 CrossRef CAS PubMed; (c) V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed.
- (a) L. Battistini, K. Bugatti, A. Sartori, C. Curti and F. Zanardi, Eur. J. Org. Chem., 2021, 2506–2528 CrossRef CAS; (b) B. Alday-Parejo, R. Stupp and C. Rüegg, Cancers, 2019, 11, 978 CrossRef CAS PubMed; (c) P. Zhong, X. Gu, R. Cheng, C. Deng, F. Meng and Z. Zhong, Int. J. Nanomed., 2017, 12, 7913–7921 CrossRef CAS PubMed; (d) D. Arosio and C. Casagrande, Adv. Drug Delivery Rev., 2016, 97, 111–143 CrossRef CAS PubMed; (e) J. S. Desgrosellier and D. A. Cheresh, Nat. Rev. Cancer, 2010, 10, 9–22 CrossRef CAS PubMed.
- (a) A. Janoniene and V. Petrikaite, Mol. Pharm., 2020, 17, 1800–1815 CrossRef CAS PubMed; (b) S. Cazzamalli, A. Dal Corso, F. Widmayer and D. Neri, J. Am. Chem. Soc., 2018, 140, 1617–1621 CrossRef CAS PubMed.
- (a) A. Rana and S. Bhatnagar, Bioorg. Chem., 2021, 112, 104946 CrossRef CAS PubMed; (b) M. Scaranti, E. Cojocaru, S. Banerjee and U. Banerji, Nat. Rev. Clin. Oncol., 2020, 17, 349–359 CrossRef PubMed; (c) J. A. Reddy, R. Dorton, A. Bloomfield, M. Nelson, C. Dircksen, M. Vetzel, P. Kleindl, H. Santhapuram, I. R. Vlahov and C. P. Leamon, Sci. Rep., 2018, 8, 8910–8943 CrossRef PubMed; (d) Y. Cao and J. Yang, Bioconjugate Chem., 2014, 25, 873–878 CrossRef CAS PubMed.
- X. Wang, A. Shirke, E. Walker, R. Sun, G. Ramamurthy, J. Wang, L. Shan, J. Mangadlao, Z. Dong, J. Li, Z. Wang, M. Schluchter, D. Luo, Y. Wang, S. Stauffer, S. Brady-Kalnay, C. Hoimes, Z. Lee and J. P. Basilion, Cancers, 2021, 13, 417 CrossRef CAS PubMed.
- (a) A. Dal Corso, ChemBioChem, 2020, 21, 3321–3322 CrossRef CAS PubMed; (b) A. Dal Corso, L. Pignataro, L. Belvisi and C. Gennari, Chem. – Eur. J., 2019, 25, 14740–14757 CrossRef CAS PubMed; (c) T. Chatzisideri, G. Leonidis and V. Sarli, Future Med. Chem., 2018, 10, 2201–2226 CrossRef CAS PubMed; (d) M. Huo, Q. Zhu, Q. Wu, T. Yin, L. Wang, L. Yin and J. Zhou, J. Pharm. Sci., 2015, 104, 2018–2028 CrossRef CAS PubMed.
- A. Alouane, R. Labruére, T. Le Saux, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2015, 54, 7492–7509 CrossRef CAS PubMed.
- (a) A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed; (b) R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS PubMed.
- V. Estrella, T. Chen, M. Lloyd, J. Wojtkowiak, H. H. Cornnell, A. Ibrahim-Hashim, K. Bailey, Y. Balagurunathan, J. M. Rothberg, B. F. Sloane, J. Johnson, R. A. Gatenby and R. J. Gillies, Cancer Res., 2013, 73, 1524–1535 CrossRef CAS PubMed.
- H. T. McMahon and E. Boucrot, Nat. Rev. Mol. Cell Biol., 2011, 12, 517–533 CrossRef CAS PubMed.
- (a) S. Chu, X. Shi, Y. Tian and F. Gao, Front. Oncol., 2022, 12 Search PubMed; (b) R. Gannimani, P. Walvekar, V. R. Naidu, T. M. Aminabhavi and T. Govender, J. Controlled Release, 2020, 328, 736–761 CrossRef CAS PubMed.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- A. Luong, T. Issarapanichkit, S. D. Kong, R. Fong and J. Yang, Org. Biomol. Chem., 2010, 8, 5105–5109 RSC.
- (a) A. Zhang, L. Yao and M. An, Chem. Commun., 2017, 53, 12826–12829 RSC; (b) S. Su, F. S. Du and Z. C. Li, Org. Biomol. Chem., 2017, 15, 8384–8392 RSC.
- S. A. Jacques, G. Leriche, M. Mosser, M. Nothisen, C. D. Muller, J.-S. Remy, A. Wagner, J. Delahousse, C. Skarbek and A. Paci, Org. Biomol. Chem., 2016, 14, 4794–4803 RSC.
- G. Leriche, M. Nothisen, N. Baumlin, C. D. Muller, D. Bagnard, J.-S. Remy, S. A. Jacques and A. Wagner, Bioconjugate Chem., 2015, 26, 1461–1465 CrossRef CAS PubMed.
- A. Abe and M. Kamiya, Bioorg. Med. Chem., 2021, 44, 116281 CrossRef CAS PubMed.
- Z. Khademi and K. Nikoofar, RSC Adv., 2020, 10, 30314–30397 RSC.
- N. Laubrock, G. Hempel, P. Schulze-Westhoff, G. Würthwein, S. Flege and J. Boos, Chromatographia, 2000, 52, 9–13 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc03052g |
| This journal is © The Royal Society of Chemistry 2022 |