
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical alkene azidocyanation via 1,4-nitrile migration†
Alex C.
Seastram
,
Mishra Deepak
Hareram
,
Thomas M. B.
Knight
and
Louis C.
Morrill
 *
*
Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: MorrillLC@cardiff.ac.uk
First published on 13th July 2022
Abstract
An electrochemical method for the azidocyanation of alkenes via 1,4-nitrile migration has been developed. This organic oxidant free method is applicable across various alkene containing cyanohydrins, and provides access to a broad range of synthetically useful 1,2-azidonitriles (28 examples). This methodology was extended to an electrochemical alkene sulfonylcyanation procedure, as well as to access a trifunctionalized hexanenitrile from a malononitrile starting material. The orthogonal derivatization of the products was also demonstrated through chemoselective transformations.
Organic azides1 are highly sought after within biological and medicinal chemistry,2 and materials science,3 both as compounds of interest and as key intermediates due to their ease of conversion into a wide range of functional groups. As such, the development of new synthetic methods to incorporate the azide functionality into organic compounds is an important and timely goal in organic synthesis.
Organic electrochemistry, which allows for the development of efficient and selective synthetic methods,4 has been established as a tool to incorporate azides into organic compounds. In 1970, Schäfer and co-workers developed a metal-free electrochemical alkene 1,2-diazidation protocol.5 This methodology was improved in terms of efficacy and selectivity by Lin and co-workers (Scheme 1A) by the introduction of MnBr2·4H2O (5 mol%) that served as a transfer reagent to deliver the azide to the alkene.6 Subsequently, Ackermann and co-workers developed a manganese-catalyzed C(sp3)-H azidation protocol under electrochemical conditions.7
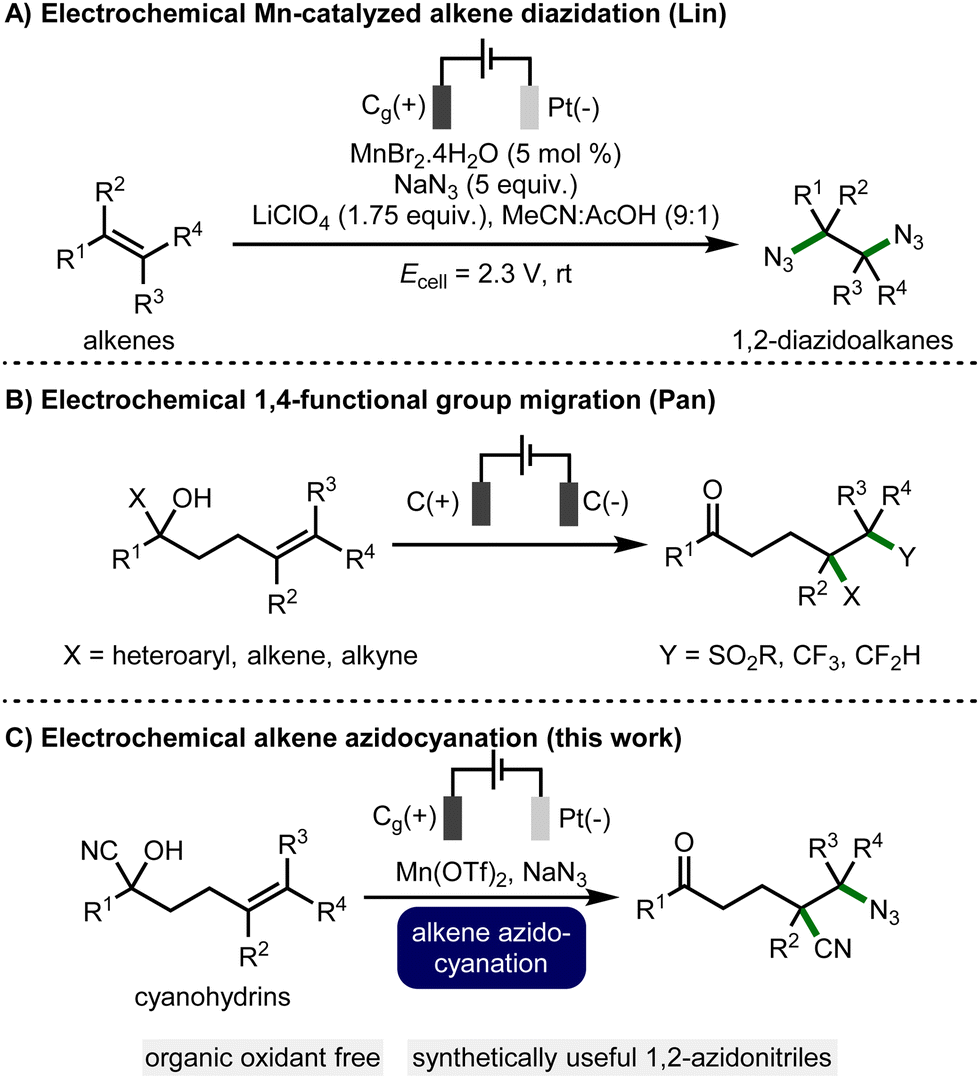 | ||
| Scheme 1 State-of-the-art and outline of the electrochemical strategy. | ||
In recent years, functional group migration has developed into a useful strategy for the difunctionalisation of unactivated alkenes.8 Through this manifold, intramolecular nitrile,9 carbonyl,10 alkynyl,10b,11 and heteroaryl9d,10b,12 migration methodologies have been developed using stoichiometric oxidants. For example, Zhu and co-workers reported an alkene azidocyanation protocol using PIDA,9a a high molecular weight oxidant which generates stoichiometric organic waste that requires separation from the desired product. To overcome the necessity for chemical oxidants, electrochemistry has been employed within this strategy to generate radical intermediates.13 Recently, Pan and co-workers reported electrochemical 1,4-migration of heteroaromatics, alkenes, and alkynes with alkene sulfonylation and fluoro-alkylation (Scheme 1B).13a,c
As such, following our research programme into the development of new electrochemical methodologies in organic synthesis, using manganese(II) salts,14 and on alkene functionalization,15 we envisaged the development of an electrochemical azidocyanation of alkenes via addition of an azide radical followed by 1,4-nitrile migration to generate a range of valuable products (Scheme 1C), and herein we report the successful realization of this approach.
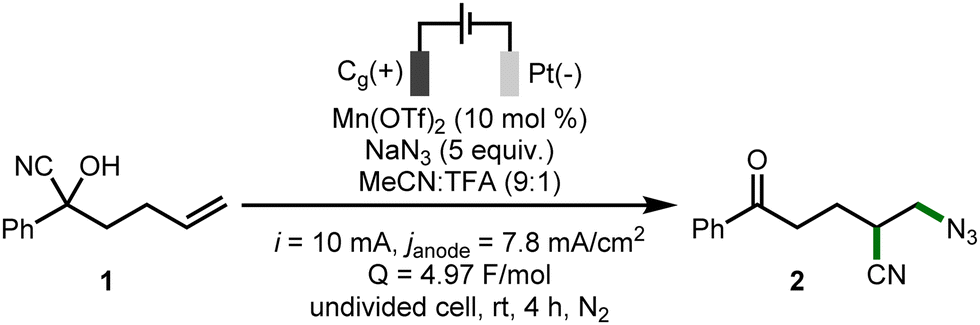
To commence our studies, cyanohydrin 1 was selected as the model substrate (Table 1).16 It was found that an electrochemical system composed of Mn(OTf)2 (10 mol%), and NaN3 (5 equiv.) as both azide source and electrolyte in MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TFA (9:1, [1] = 0.05 M) using galvanostatic conditions (i = 10 mA, janode = 7.8 mA cm−2, Q = 4.97 F mol−1), a graphite anode and a platinum cathode at r.t. for 4 h under N2, enabled the azido-cyanation of 1, with ketone 2 formed in a 55% NMR yield (entry 1). No conversion occurs in the absence of electricity (entry 2), and the yield of 2 was significantly reduced in the absence of Mn(OTf)2 (entry 3). Employing a constant cell potential (Ecell = 2.4 V) saw 2 obtained in a 55% yield, albeit in a significantly increased reaction time (entry 4). Alterations in the current applied (i = 12.5 mA or 7.5 mA) lowered the yield of 2 (entries 5 and 6), as did adding LiClO4 (1.75 equiv.) as a supporting electrolyte (entry 7) and substituting the platinum foil cathode for graphite or Ni plate (entries 8 and 9). Employing MnBr2·4H2O or Mn(OAc)2·4H2O in place of Mn(OTf)2 was also detrimental to conversion (entries 10 and 11). Substituting TFA for AcOH as the proton source resulted in a reduced conversion to 2 (entry 12), presumably due to the decreased solubility of NaN3 in the MeCN:AcOH solvent system. It was found that lowering the quantity of NaN3 to 2.5 equivalents reduced the NMR yield of 2 to 19% (entry 13), but reducing the loading of Mn(OTf)2 to 5 mol% had a negligible impact on the yield obtained (entry 14). Extending the reaction time to 5 h saw a decreased NMR yield of 2 to 42% (entry 15), suggesting that the product may be somewhat unstable under the reaction conditions. Evidence supporting this claim was obtained when 2 was subjected to the “standard” reaction conditions, where only 64% was returned by 1H NMR analysis of the crude reaction mixture.
:TFA (9:1, [1] = 0.05 M) using galvanostatic conditions (i = 10 mA, janode = 7.8 mA cm−2, Q = 4.97 F mol−1), a graphite anode and a platinum cathode at r.t. for 4 h under N2, enabled the azido-cyanation of 1, with ketone 2 formed in a 55% NMR yield (entry 1). No conversion occurs in the absence of electricity (entry 2), and the yield of 2 was significantly reduced in the absence of Mn(OTf)2 (entry 3). Employing a constant cell potential (Ecell = 2.4 V) saw 2 obtained in a 55% yield, albeit in a significantly increased reaction time (entry 4). Alterations in the current applied (i = 12.5 mA or 7.5 mA) lowered the yield of 2 (entries 5 and 6), as did adding LiClO4 (1.75 equiv.) as a supporting electrolyte (entry 7) and substituting the platinum foil cathode for graphite or Ni plate (entries 8 and 9). Employing MnBr2·4H2O or Mn(OAc)2·4H2O in place of Mn(OTf)2 was also detrimental to conversion (entries 10 and 11). Substituting TFA for AcOH as the proton source resulted in a reduced conversion to 2 (entry 12), presumably due to the decreased solubility of NaN3 in the MeCN:AcOH solvent system. It was found that lowering the quantity of NaN3 to 2.5 equivalents reduced the NMR yield of 2 to 19% (entry 13), but reducing the loading of Mn(OTf)2 to 5 mol% had a negligible impact on the yield obtained (entry 14). Extending the reaction time to 5 h saw a decreased NMR yield of 2 to 42% (entry 15), suggesting that the product may be somewhat unstable under the reaction conditions. Evidence supporting this claim was obtained when 2 was subjected to the “standard” reaction conditions, where only 64% was returned by 1H NMR analysis of the crude reaction mixture.
|
|
|||
|---|---|---|---|
| Entry | Variation from “standard” conditions | 1 (%) | 2 (%) |
| a Reactions performed using 0.3 mmol of cyanohydrin 1 using the ElectraSyn 2.0 batch electrochemical reactor. [1] = 0.05 M. b Yield after 4 h as determined by 1H NMR analysis of the crude reaction mixture with 1,3,5-trimethylbenzene as the internal standard. Isolated yield given in parentheses. c 192 minutes reaction time. d 320 minutes reaction time. e 300 minutes reaction time. | |||
| 1 | None | <2 | 55 (51) |
| 2 | No electricity | 94 | <2 |
| 3 | No Mn(OTf)2 | 34 | 23 |
| 4 | E cell = 2.4 V | <2 | 55 |
| 5c | i = 12.5 mA, janode = 9.8 mA cm−2 | 4 | 32 |
| 6d | i = 7.5 mA, janode = 5.9 mA cm−2 | <2 | 42 |
| 7 | LiClO4 (1.75 equiv.) as electrolyte | 20 | 38 |
| 8 | Graphite cathode instead of Pt foil | 6 | 23 |
| 9 | Ni plate cathode instead of Pt foil | 10 | 43 |
| 10 | MnBr2·4H2O instead of Mn(OTf)2 | <2 | 34 |
| 11 | Mn(OAc)2·4H2O instead of Mn(OTf)2 | <2 | 48 |
| 12 | AcOH instead of TFA | 20 | 30 |
| 13 | NaN3 (2.5 equiv.) | 29 | 19 |
| 14 | Mn(OTf)2 (5 mol%) | 8 | 50 |
| 15e | Q = 6.22 F mol−1 | <2 | 42 |
With optimized reaction conditions in hand (Table 1, entry 1), the full scope of the electrosynthetic method with respect to the cyanohydrin starting materials was evaluated (Scheme 2). Upon increasing the reaction scale to 1.0 mmol, product 2 was obtained in a 64% isolated yield. Employing cyanohydrin starting materials, it was found that steric hinderance around the aromatic ring did not impact the reaction in an appreciable manner, with 4-, 3-, and 2-Me substitution providing access to the corresponding products (products 3–5) in comparable yields. Various substituents could be incorporated into the aromatic ring, including aryl (e.g., 4-Ph), and electron-withdrawing groups (e.g., 4-CN), with the corresponding azido-nitrile products accessed in moderate to good yields (products 6–8). Halogens could be tolerated in the para position, as could pinacol-borane (products 9–13), giving the potential for further derivatization of the products obtained. The electrochemical method exhibits good functional group tolerance, demonstrated by the presence of carboxylic acid, methyl ester, primary amide, tertiary amide, and ketal in the 4-position (products 14–18). Methyl ester 16 was obtained in a 75% isolated yield, suggesting that this electron-withdrawing substituent may stabilise intermediates in this reaction, disfavouring non-productive pathways that may occur under the electrochemical conditions. Extension of the aromatic system was tolerated, with 1-naphthyl product 19 obtained in a 38% NMR yield. Alkyl cyanohydrins were also evaluated under the reaction conditions (e.g., Bn), which provided the products 20–24 in moderate yields. Substitution on the olefin was then evaluated, allowing for the formation of secondary azides 25 and 26 (as a single diastereoisomer), as well as quaternary nitrile 27. Substitution on the alkyl chain allowed for the formation of tetralone 28 in a 51% NMR yield. Substrates with decreased and increased distances between the cyanohydrin and alkene resulted in no observable product formation. Alternative transformations were also evaluated under this electrosynthetic manifold, with sulfone 29 formed from 1 by substituting the NaN3 for NaSO2Ph, demonstrating the ability to initiate the nitrile migration via addition of sulfonyl radicals to the alkene. Malononitrile 30 was also successfully converted to trifunctionalized hexanenitrile 31 in a 42% NMR yield when subjected to the “standard” reaction conditions (Table 1, entry 1).
 | ||
| Scheme 2 Scope. Reactions performed using 0.3 mmol of starting material using the ElectraSyn 2.0 batch electrochemical reactor. [Substrate] = 0.05 M. All yields are as determined by 1H NMR analysis of the crude reaction mixture with 1,3,5-trimethylbenzene as the internal standard. Isolated yield after chromatographic purification given in parentheses. a1.0 mmol, t = 13 h 20 min. b5 h (6.22 F mol−1). cAs determined by 1H NMR analysis of the crude reaction mixture. dNaSO2Ph (5 equiv.) instead of NaN3. | ||
To demonstrate product utility, the orthogonal functionalization of azido-nitrile 2 was investigated (Scheme 3). It was found that the azide could be converted to the corresponding 1,4-disubstituted 1,2,3-triazole 32 in a 47% isolated yield via a CuAAC reaction with phenylacetylene.17 Furthermore, the ketone was converted to hydrazone 33via condensation with benzhydrazide in an 88% isolated yield.18
 | ||
| Scheme 3 Product utility. | ||
To obtain insight into the reaction mechanism, cyanohydrin 34 was subjected to the optimized electrochemical reaction conditions as a radical clock mechanistic probe (Scheme 4A). It was found that ring-opened azido-nitrile 35 was obtained as the sole product in a 56% NMR yield, signifying the presence of radical intermediates in the reaction mechanism. Cyclic voltammetry was employed to gain further insight into the mechanism of this electrochemical process.16 In accordance with the literature,6,14 Mn(OTf)2 displays no redox events between 0 and 1.6 V vs. Fc/Fc+, and TBAN3 exhibits an irreversible oxidation event at ∼0.97 V vs. Fc/Fc+, indicating the formation of azide radicals, which can themselves induce the desired reactivity (Table 1, entry 3). However, the combination of Mn(OTf)2 with TBAN3 produced a new irreversible oxidation event at ∼0.50 V vs. Fc/Fc+, providing evidence for the generation of a Mn(III)X2N3 species from [Mn(II)X2N3]−.
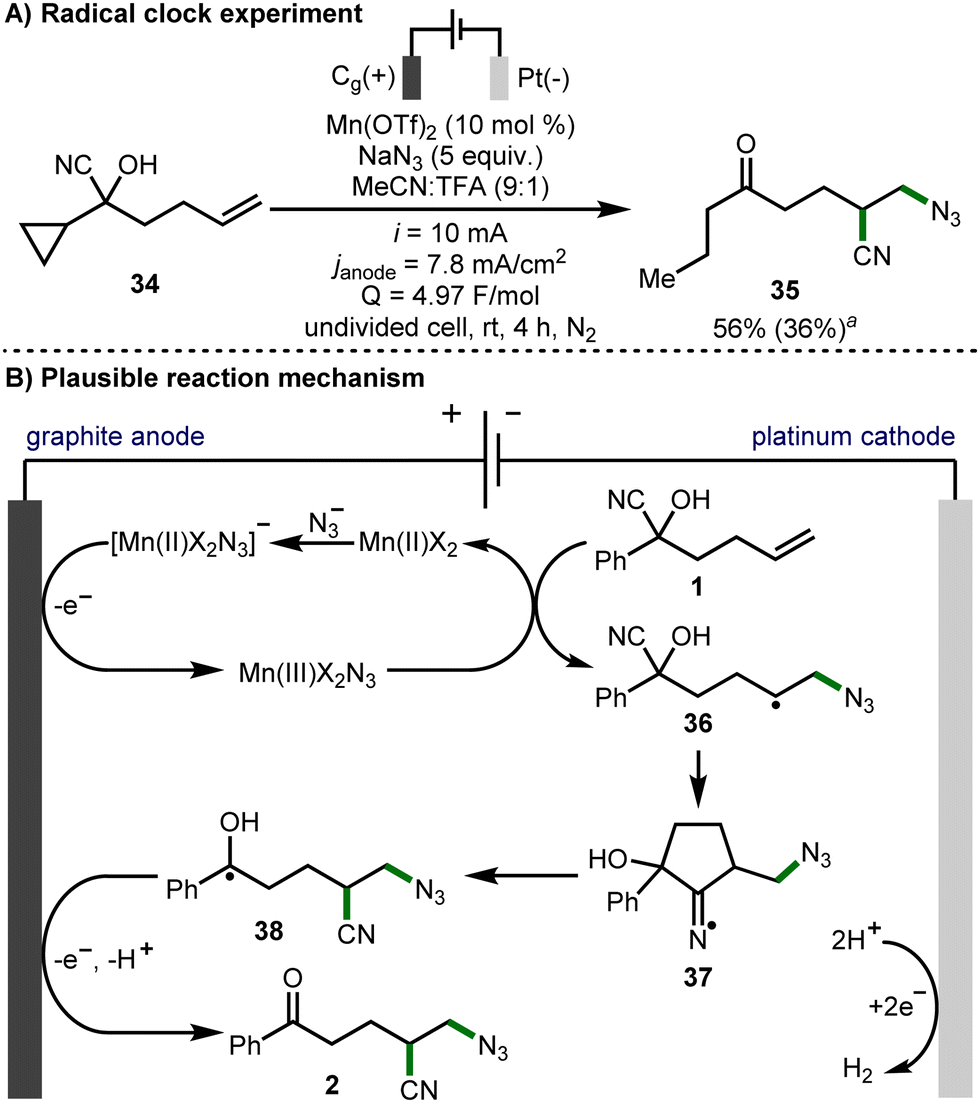 | ||
| Scheme 4 Reaction mechanism. aAs determined by 1H NMR analysis of the crude reaction mixture. Isolated yield in parentheses. | ||
With these observations, as well as literature precedent from previous related investigations,6,9a a plausible reaction mechanism would initiate with the formation of [Mn(II)X2N3]− from Mn(II)X2 and NaN3, which is oxidized at the anode to form Mn(III)X2N3 (Scheme 4B). This intermediate delivers an azide radical to cyanohydrin 1, giving secondary alkyl radical intermediate 36. Intramolecular interception of this secondary alkyl radical by the nitrile affords cyclic iminyl radical 37, which can then undergo β-scission to afford α-hydroxy alkyl radical 38. A second oxidation at the anode, followed by proton loss affords product 2. Hydrogen gas is evolved via proton reduction at the cathode.
In conclusion, we have developed a new electrochemical method for the azidocyanation of alkenes. The method is applicable across various alkene containing cyanohydrins, providing access to a broad range of synthetically useful 1,2-azidonitriles (28 examples). The methodology was also extended to an electrochemical sulfonylcyanation procedure, as well as to a malononitrile starting material, giving access to a trifunctionalized hexanenitrile product. The orthogonal derivatization of the products was also demonstrated through chemoselective transformations.
The data that support the findings of this study are openly available in the Cardiff University data catalogue at http://doi.org/10.17035/d.2022.0214744028.
We gratefully acknowledge the School of Chemistry, Cardiff University for generous support, the EPSRC for a doctoral training grant for a PhD studentship (A.S., EP/R513003/1), and a standard research grant (M. D. H., EP/R006504/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) N. Jung and S. Bräse, Angew. Chem., Int. Ed., 2012, 51, 12169–12171 CrossRef CAS PubMed; (b) S. Chiba, Synlett, 2012, 21–44 CrossRef CAS; (c) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- For selected reviews, see: (a) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed; (b) M. D. Best, Biochemistry, 2009, 48, 6571–6584 CrossRef CAS PubMed ; For selected papers, see: ; (c) P. P. Geurink, W. A. Van Der Linden, A. C. Mirabella, N. Gallastegui, G. De Bruin, A. E. M. Blom, M. J. Voges, E. D. Mock, B. I. Florea, G. A. Van Der Marel, C. Driessen, M. Van Der Stelt, M. Groll, H. S. Overkleeft and A. F. Kisselev, J. Med. Chem., 2013, 56, 1262–1275 CrossRef CAS PubMed; (d) D. B. Smith, G. Kalayanov, C. Sund, A. Winqvist, T. Maltseva, V. J. P. Leveque, S. Rajyaguru, S. Le Pogam, I. Najera, K. Benkestock, X. X. Zhou, A. C. Kaiser, H. Maag, N. Cammack, J. A. Martin, S. Swallow, N. G. Johansson, K. Klumpp and M. Smith, J. Med. Chem., 2009, 52, 2971–2978 CrossRef CAS PubMed.
- W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572–2590 CrossRef CAS.
- For selected reviews, see: (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (b) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed; (c) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (d) K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed; (e) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed; (f) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- H. Schäfer, Angew. Chem., Int. Ed. Engl., 1970, 9, 158–159 CrossRef.
- N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed.
- T. H. Meyer, R. C. Samanta, A. Del Vecchio and L. Ackermann, Chem. Sci., 2021, 12, 2890–2897 RSC.
- X. Wu and C. Zhu, Acc. Chem. Res., 2020, 53, 1620–1636 CrossRef CAS PubMed.
- (a) Z. Wu, R. Ren and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 10821–10824 CrossRef CAS PubMed; (b) M. Ji, Z. Wu, J. Yu, X. Wan and C. Zhu, Adv. Synth. Catal., 2017, 359, 1959–1962 CrossRef CAS; (c) R. Ren, Z. Wu, L. Huan and C. Zhu, Adv. Synth. Catal., 2017, 359, 3052–3056 CrossRef CAS; (d) D. Chen, Z. Wu, Y. Yao and C. Zhu, Org. Chem. Front., 2018, 5, 2370–2374 RSC; (e) M. Ji, Z. Wu and C. Zhu, Chem. Commun., 2019, 55, 2368–2371 RSC; (f) C. Chang, H. Zhang, X. Wu and C. Zhu, Chem. Commun., 2022, 58, 1005–1008 RSC.
- (a) Z. L. Li, X. H. Li, N. Wang, N. Y. Yang and X. Y. Liu, Angew. Chem., Int. Ed., 2016, 55, 15100–15104 CrossRef CAS PubMed; (b) J. Yu, D. Wang, Y. Xu, Z. Wu and C. Zhu, Adv. Synth. Catal., 2018, 360, 744–750 CrossRef CAS.
- (a) X. Tang and A. Studer, Chem. Sci., 2017, 8, 6888–6892 RSC; (b) Y. Xu, Z. Wu, J. Jiang, Z. Ke and C. Zhu, Angew. Chem., Int. Ed., 2017, 56, 4545–4548 CrossRef CAS PubMed.
- (a) Z. Wu, D. Wang, Y. Liu, L. Huan and C. Zhu, J. Am. Chem. Soc., 2017, 139, 1388–1391 CrossRef CAS PubMed; (b) M. Wang, Z. Wu, B. Zhang and C. Zhu, Org. Chem. Front., 2018, 5, 1896–1899 RSC; (c) X. J. Wei and T. Noël, J. Org. Chem., 2018, 83, 11377–11384 CrossRef CAS PubMed; (d) H. Zhang, X. Wu, Q. Zhao and C. Zhu, Chem. – Asian J., 2018, 13, 2453–2457 CrossRef CAS PubMed; (e) N. Tang, S. Yang, X. Wu and C. Zhu, Tetrahedron, 2019, 75, 1639–1646 CrossRef CAS.
- (a) Y. Gao, H. Mei, J. Han and Y. Pan, Chem. – Eur. J., 2018, 24, 17205–17209 CrossRef CAS PubMed; (b) M. W. Zheng, X. Yuan, Y. S. Cui, J. K. Qiu, G. Li and K. Guo, Org. Lett., 2018, 20, 7784–7789 CrossRef CAS PubMed; (c) Z. Zou, W. Zhang, Y. Wang, L. Kong, G. Karotsis, Y. Wang and Y. Pan, Org. Lett., 2019, 21, 1857–1862 CrossRef CAS PubMed; (d) C. Chen, J. C. Kang, C. Mao, J. W. Dong, Y. Y. Xie, T. M. Ding, Y. Q. Tu, Z. M. Chen and S. Y. Zhang, Green Chem., 2019, 21, 4014–4019 RSC; (e) H. I. Jung, Y. Kim and D. Y. Kim, Org. Biomol. Chem., 2019, 17, 3319–3323 RSC; (f) Y. J. Kim and D. Y. Kim, Org. Lett., 2019, 21, 1021–1025 CrossRef CAS PubMed; (g) J. C. Kang, Y. Q. Tu, J. W. Dong, C. Chen, J. Zhou, T. M. Ding, J. T. Zai, Z. M. Chen and S. Y. Zhang, Org. Lett., 2019, 21, 2536–2540 CrossRef CAS PubMed; (h) Z. Guan, H. Wang, Y. Huang, Y. Wang, S. Wang and A. Lei, Org. Lett., 2019, 21, 4619–4622 CrossRef CAS PubMed.
- B. D. W. Allen, M. D. Hareram, A. C. Seastram, T. McBride, T. Wirth, D. L. Browne and L. C. Morrill, Org. Lett., 2019, 21, 9241–9246 CrossRef CAS PubMed.
- J. Harnedy, M. D. Hareram, G. J. Tizzard, S. J. Coles and L. C. Morrill, Chem. Commun., 2021, 57, 12643–12646 RSC.
- See the ESI† for full experimental details.
- X. Creary, A. Anderson, C. Brophy, F. Crowell and Z. Funk, J. Org. Chem., 2012, 77, 8756–8761 CrossRef CAS PubMed.
- X. L. Yang, X. X. Peng, F. Chen and B. Han, Org. Lett., 2016, 18, 2070–2073 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization data, experimental procedures, characterization of new compounds and spectral data. See DOI: https://doi.org/10.1039/d2cc02958h |
| This journal is © The Royal Society of Chemistry 2022 |