
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphinoborinium cation: a synthon for cationic B–P bond systems†
Kinga
Kaniewska-Laskowska
 ,
Katarzyna
Klimsiak
,
Natalia
Szynkiewicz
,
Jarosław
Chojnacki
and
Rafał
Grubba
*
,
Katarzyna
Klimsiak
,
Natalia
Szynkiewicz
,
Jarosław
Chojnacki
and
Rafał
Grubba
*
Department of Inorganic Chemistry, Faculty of Chemistry and Advanced Materials Center, Gdańsk University of Technology, ul. Narutowicza 11/12, 80-233 Gdańsk, Poland. E-mail: rafal.grubba@pg.edu.pl
First published on 12th August 2022
Abstract
Herein, we report access to phosphinoborinium cations via heterolytic cleavage of the boron–bromide bond in bromophosphinoborane. The product of the reaction was isolated as a dimeric dication possessing a planar B2P2 core. Activation of the C–H and C–P bonds in the dication led to the formation of the borinium–phosphaborene adduct. Reactivity studies revealed that the title cation exhibits ambiphilic properties and intramolecular frustrated Lewis pair features.
Due to Lewis acidic properties resulting from the empty p-orbital and coordinative unsaturation of the B atom, tri-coordinate, neutral boron compounds play key roles in nonmetallic catalysis.1 The discovery of frustrated Lewis pairs (FLPs)2 has stimulated the rapid development of the chemistry of triorganoboranes,3 which are most commonly the Lewis acidic components of FLPs capable of activating strong chemical bonds in simple inorganic and organic molecules.4–6
Low-coordinate boronic cations are even more electron deficient than neutral boron Lewis acids. Generally, three main types of boronic cations can be distinguished by taking into account the coordination number of boron: borinium [R2B]+, borenium [R2BL]+, and boronium [R2BL2]+ ions.7,8 The first two are attractive synthetic targets owing to their strong Lewis acidic properties. In the past, borinium and borenium cations were only considered chemical curiosities; however, the development of modern synthetic methods allowed isolation and characterization of these species in the condensed phase. Due to the high reactivities of low-coordinate boronic cations, their syntheses are not trivial and require the application of electronic and/or steric stabilization. The first borinium cation was obtained by Nöth and coworkers,9,10 and electron-donating amino groups were introduced at the B-centre. Another milestone in this area was the synthesis and isolation of [Mes2B]+ by the Shoji group.11–13 These results showed that the presence of π-donating ligands was not necessary for the stabilization of the borinium cations. Stephan and coworkers used two phosphoraniminato ligands for the stabilization of boron centres with a low coordination number;14 moreover, they showed that Nöth's and Shoji's cations exhibited high reactivity towards small inorganic and organic molecules.15,16 The recent development of boronic cation chemistry confirms the applicability of these reactive species in stoichiometric reactions and as catalysts for hydrogenation, hydrosilylation, hydroboration, C–H bond borylation, and C–C bond hydrogenolysis reactions.17–23
Our research interests are focused on the chemistry of low-coordinate B–P bond systems such as phosphinoboranes,24 diphosphinoboranes,25 and triphosphinoboranes.26 Despite the presence of one to three B–P bonds, these systems retain Lewis acidic and Lewis basic character of the reactive boron and phosphorus centres and behave as intramolecular FLPs to activate H2, CO2, other heterocumulenes, SO2, N2O, and BH3.24,26–28 As a continuation of our previous studies, we decided to introduce this research concept to cationic boron compounds and synthesize low-coordinate boronic cations possessing B–P functionality.
As a precursor to the phosphinoborinium cation, we utilized Cy2N(Br)BPtBuPh (1), which was obtained via a simple two-step reaction of BBr3 with Cy2NH followed by the addition of tBuPhPLi. In a recent study, we used analogous amino(bromo)phosphinoboranes for syntheses of diphosphinoboranes with mixed phosphanyl substituents.25 Spectroscopic and X-ray diffraction data for 1 are presented in the ESI.† As a source of weakly coordinating anions (WCAs), we chose Krossing's salt Li[Al(OC(CF3)3)4] due to its synthetic accessibility and chemical inertness.29 According to 31P and 11B spectroscopic analyses, an equimolar reaction of 1 with the aforementioned salt in dichloromethane (DCM) led to the formation of three ionic species, A[WCA]2, B[WCA], and C[WCA], in a molar ratio ≈ 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1; this was accompanied by precipitation of LiBr (Scheme 1).
:1:1; this was accompanied by precipitation of LiBr (Scheme 1).
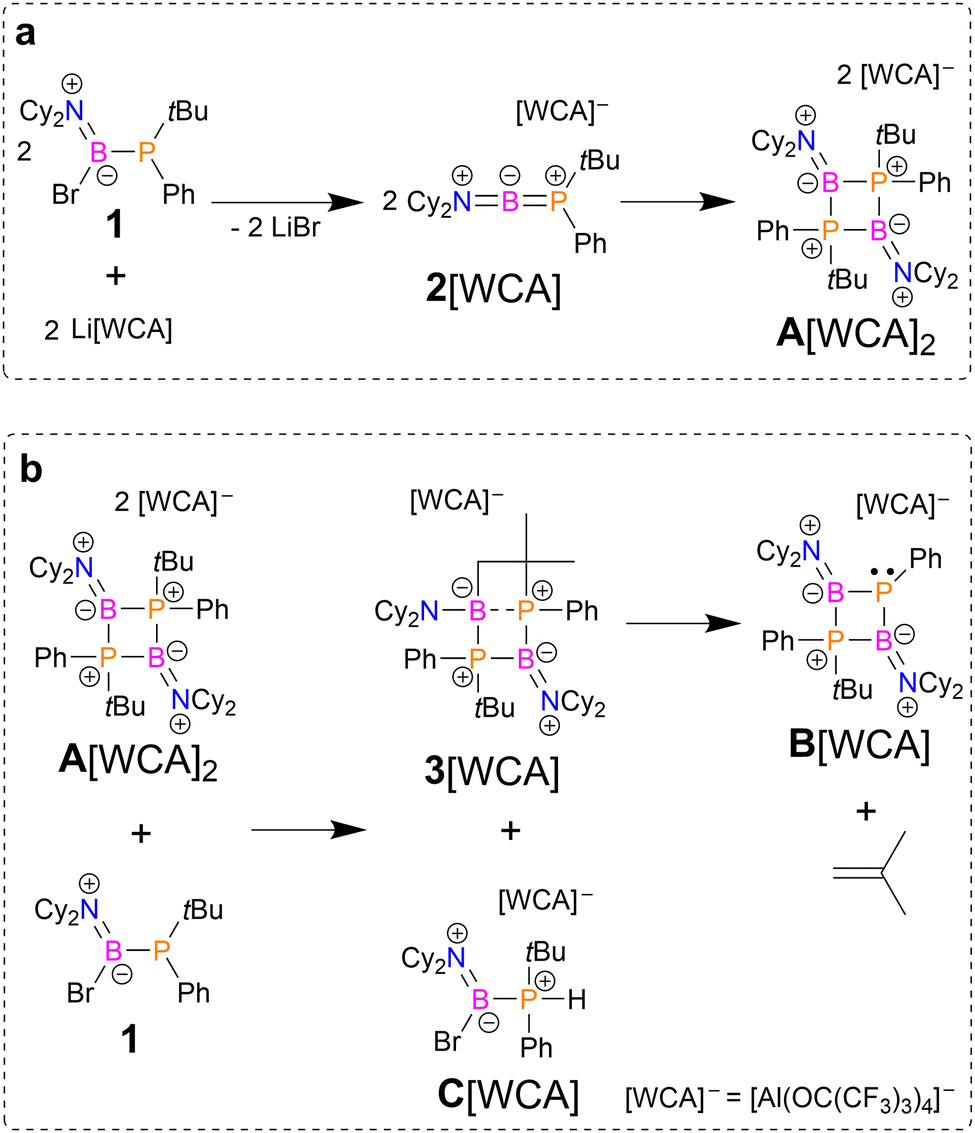 | ||
| Scheme 1 Syntheses of cations A2+, B+, and C+ (synthetic details: solvent CH2Cl2; temperature from −30 °C to 25 °C; yields: A[WCA]2 – 32%, B[WCA] – 61%. The bulk purity of A[WCA]2 and B[WCA] was confirmed by elemental analysis). | ||
Additionally, the evolution of gas bubbles was observed during the reaction, especially in the case of pressure equalization between the reaction vessel and the argon manifold of the Schlenk line. This gaseous product together with the solvent was collected in a liquid nitrogen trap and unambiguously identified as isobutene by 1H and 13C NMR spectroscopy (Fig. S41 and S42, ESI†).
Due to differences in the solubilities of the reaction products in hydrocarbons, they can be separated by fractional crystallization. Colourless crystals of A[WCA]2 were deposited from a DCM solution at 4 °C. Collectively, NMR spectroscopic and X-ray diffraction studies revealed that the obtained salt consisted of dication A2+ (diphosphoniodiborane), which can be seen as the dimer of the monomeric phosphinoborinium cation 2+ (Scheme 1a). Indeed, a calculation of the Gibbs free energy for dimerization of 2+ in a DCM solution showed that this reaction is strongly thermodynamically favoured (ΔG0298K = −29.7 kcal mol−1).30 The 11B and 31P{1H} NMR spectra of A2+ displayed very broad signals at 36.0 ppm and 11.7 ppm, respectively. Due to the broadness of the signals, boron–phosphorus coupling was not observed. The 11B chemical shift indicated a low coordination number for the boron atom. Moreover, the 19F and 27Al NMR spectra showed characteristic signals for [Al(OC(CF3)3)4]−, confirming that this counterion remained untouched. The molecular structure of A2+ is presented in Fig. 1. The most characteristic structural feature of A2+ is the planar B2P2 core. The geometries around the nitrogen and boron atoms are trigonal planar with the sums of angles very close to 360°. The P atoms exhibit distorted tetrahedral geometries.
 | ||
| Fig. 1 X-Ray structures of cations A2+ and B+. The counterions and H-atoms are omitted for clarity. The thermal ellipsoids are shown at the 50% probability level. | ||
The relatively short N1–B1 distance of 1.377(6) Å together with the planar geometry around the N1 and B1 atoms indicates significant π-interactions occurring between these atoms. The B1–P1 (1.979(4) Å) and B1′–P1′ (1.981(5) Å) distances are in the typical range for single B–P bonds (1.96 Å).31 The tBu and Ph substituents are located in trans positions with respect to the planar B2P2 ring. Interestingly, signals attributable to the cis isomer were found in the NMR spectra of isolated crystals of A[WCA]2. The molar ratio of the trans and cis isomers was calculated as 84:16 based on the integration of the well-separated CH signals for Cy groups in the 1H NMR spectrum. Furthermore, DFT calculations showed that the trans isomer has a lower energy than the cis isomer, and the free energy difference is −7.4 kcal mol−1 in a DCM solution.30
The electronic structure of A2+ was investigated by theoretical methods.32 Natural population analysis (NPA) revealed high positive charges located at the phosphorus and boron atoms of the central planar core with values of +0.866 and +0.512, respectively. The B–N bonds are significantly polarized with a high negative charge located at the nitrogen atom (−0.741). The Wiberg bond index (WBI) calculated for B1–P1 and B1′–P1′ (0.904, 0.914) confirmed the single bond character of these bonds. The WBI for B1–N1/B1′–N1′ was 1.196, which is consistent with the short boron–nitrogen distance and partial multiple bond character.32 The LUMO of A2+ is delocalized over the boron and phosphorus atoms and is composed of two lobes separated by a nodal plane corresponding to the B2P2 plane. The HOMO of A2+ exhibits electron density localized between the B and P atoms (σ-bonding) with a contribution from electron density localized on phenyl substituents (Fig. S54, ESI†).32
To our knowledge, there are no reports of planar dicationic B2P2 cyclic systems containing tri-coordinate boron and four-coordinate phosphorus atoms. Analogous planar B2P2 cores can be found only in very few neutral species, such as the stable diradical (tBuBPiPr2)2 reported by the Bertrand group.33 Very recently, Braunschweig and coworkers described the synthesis of a neutral, planar NHC-stabilized B2P2 ring via a reaction of diboryne B2(SIDep)2 (SIDep = 1,3-bis(2,6-diethylphenyl)imidazolin-2-ylidene) with a diphosphane (Et2P)2.34 Oxidation of the resulting compound led to the formation of a dication possessing a butterfly structure and a B–B bond,34 which is in strong contrast with the structural features of A2+ described above.
After the separation of the A[WCA]2 crystals, the DCM solution was layered with pentane and stored at +4 °C. Diffusion crystallization gave large, colourless crystals of B[WCA]. According to NMR spectroscopic and X-ray diffraction studies, B+ can be described formally as an adduct of borinium cation 2+ and the phosphaborene PhPBNiPr2. Cooling the mother liquor to −20 °C gave a mixture of colourless crystals of B[WCA] and C[WCA]. The X-ray study revealed that the latter compound contains a C+ borenium cation resulting from protonation of starting 1 (Fig. S49, ESI†). The presence of the P–H functional group in the structure of C+ was confirmed both by analysis of the Fourier density map and 1JPH coupling with the expected large absolute value of 346.9 Hz (Fig. S40, ESI†). The salt C[WCA] decomposes in solution to tBuPhPH and Cy2NBBr2 which prevented its separation and full characterization (Scheme S5, ESI†). These experimental observations suggested that B+ was formed via the reaction of A2+ with phosphinoborane 1. This assumption was additionally confirmed by two independent experiments. First, the reaction of Li[WCA] with an excess of 1 (molar ratio 1:1.5) did not lead to the formation of A2+ but gave two boronic cations, B+ and C+. Similar to the initial, equimolar reaction, precipitation of LiBr and evolution of isobutene gas were observed. Second, we added 1 to a solution of the previously isolated crystals A[WCA]2 (molar ratio 1:1), which yielded only B[WCA], C[WCA], and isobutene. Considering all of these experimental observations, we propose a plausible reaction path leading to cation B+ (Scheme 1b). In the first step, A2+ is deprotonated by 1, which leads to the formation of short-lived intermediate 3+ and borenium cation C+. The proposed structure for 3+ derived from theoretical studies shows that deprotonation of one methyl group of A2+ leads to the formation of a new bond between the deprotonated C atom and the electrophilic B atom and a significant weakening of one B–P bond (Fig. S58, ESI†). In the next step, 3+ eliminates isobutene as a result of C–B and C–P bond activation, yielding B+. DFT calculations including solvation in DCM revealed that the first step of this reaction, which involved the formation of unstable 3+, is only slightly endergonic, whereas the second step is strongly exergonic; the ΔGo298K values were 2.4 kcal mol−1 and −24.6 kcal mol−1, respectively.30 Very recently, Stephan and coworkers found that the borinium cation [B(N(SiMe3)2)2]+ can act as a source of a proton,35 which is in line with our results.
The unprecedented structure of B+ was investigated by spectroscopic, diffraction, and theoretical methods. The 31P{H} NMR spectrum of B+ consisted of two broad doublets at 6.5 ppm and −17.3 ppm with large 2JPP coupling (391 Hz), which corroborated the presence of two inequivalent P-centres. Similar to A2+, only one broad signal was visible in the 11B spectrum of B+, and it exhibited a chemical shift characteristic of a tri-coordinate boron centre (35.1 ppm). The solid-state structure of B+ is depicted in Fig. 1. In contrast to the structure of A2+, the P1 atom is tri-coordinate and bonded to the B1, B2, and C1 atoms of the phenyl group. Otherwise, the P2 atom retained the tetrahedral geometry, as observed for the phosphorus atoms in A2+. Interestingly, the geometry around P1 exhibited a high degree of planarity, and the angles summed to 349.38°. Furthermore, the structure of B+ exhibited relatively short B1–P1 and B2–P1 distances with values of 1.881(2) Å and 1.882(2) Å, respectively. The flattened geometry at the P1 atom together with the significant shortening of the B1–P1 and B2–P1 bonds suggested the presence of π-interactions resulting from the overlap of the lone pair electrons on the P1 atom with the formally empty p-orbitals of the B1 and B2 atoms. The N1–B1 (1.376(2) Å), N2–B2 (1.380(2) Å), B1–P2 (1.970(2) Å) and B2–P2 (1.963(2) Å) bond lengths were very similar to the B–N and B–P distances observed for A2+. Moreover, a comparison of the A2+ and B+ structures revealed common planar geometries around the boron and nitrogen atoms. The B2P2 ring in B+ was not planar, as observed for A2+, but folded along the P1–P2 vector, and the angle between the B1P1B2 and B1P2B2 planes was 155.65°.
DFT calculations of the frontier molecular orbitals of B+ revealed that the HOMO and LUMO represented mainly B1–P1–B2 π-bonding and π*-antibonding orbitals, respectively (Fig. S55, ESI†).32 The WBI indexes calculated for B1–P1 (1.167) and B2–P1 (1.176) were in accordance with the partial multiple bond character of these bonds. On the other hand, the single bond character of B1–P2 and B2–P2 was confirmed by WBIs with values of 0.915 and 0.926, respectively. The WBIs for the boron–nitrogen bonds in B+ were slightly smaller than that for A2+ (1.118–1.132 vs. 1.196).32 A NPA analysis showed that all atoms of the B2P2 ring were positively charged; the highest positive charge was found at the P2 atom (0.977), and significantly lower positive charges were found at P1 (0.379) and the boron atoms (0.307). As with A2+, the highest negative charges were located at the nitrogen atoms (−0.733, −0744).32 These results showed that despite the intrinsic electron deficiency of B+, this cation also has the feature of a Lewis base (the electron pair at the P1 atom).
We tested the applicability of phosphinoborinium cation in the activation of small molecules. The cation 2+ generated in situ reacted with benzophenone giving adduct D[WCA] as only one product (Scheme 2). The 11B and 31P{1H} NMR spectra of this compound display very broad signals at 29.3 ppm and 68.4 ppm, respectively, which corroborate with the B–P bond and tricoordinate B atom occurring in the structure. The colourless crystals of D[WCA] were isolated from DCM solution at −20 °C. The X-ray diffraction studies shed more light on the structural features of D+ (Scheme 2 and Fig. S50, Table S1, ESI†). The adduct D+ possesses a planar P1–B1–O1–C1 core resulting from electron donor–acceptor interactions between the phosphinoborinium cation and benzophenone. The Lewis basic P1 atom acts as an electron pair donor to the C1 atom of the carbonyl group, whereas the low-coordinate Lewis acidic B1 atom accepts an electron pair from the O1 atom of benzophenone. These observations confirmed the ambiphilic properties of the phosphinoborinium cation, which exhibits characteristic reactivity for intramolecular frustrated Lewis pairs. Interestingly, in the case of ambiphilic species containing tri-coordinate B atoms, such as phosphinoboranes and diphosphinoboranes, the small molecules are inserted into the B–P bond (except H2).24,27,28,36 Similar to the product of the reaction of the phosphinoborinium cation with benzophenone, the B–P bond is retained in adducts of neutral phosphaborene (containing di-coordinate B atom) with ketones, which were further utilized in phospha-bora-Wittig reaction.37
 | ||
| Scheme 2 Synthesis and X-ray structure of cation D+ (synthetic details: solvent CH2Cl2; temperature from −50 °C to 25 °C; yield 40%. The bulk purity of D[WCA] was confirmed by elemental analysis. The counterion and H-atoms are omitted for clarity. The thermal ellipsoids are shown at the 50% probability level). | ||
In conclusion, we have provided a simple method for the generation of phosphinoborinium cation intermediates utilizing synthetically accessible bromophosphinoboranes. The obtained borinium cations can act as building blocks for cationic frameworks possessing B–P functionality. The presented results open a new chapter in the chemistry of low-coordinate boronic cations by giving access to species that combine the features of both Lewis acids and Lewis bases. Having a simple method for syntheses of phosphinoborinium cations available, we are currently working on applications of these species in stoichiometric and catalytic transformations that allow activation of strong chemical bonds.
Financial support of these studies from Gdańsk University of Technology by the DEC-3/2020/IDUB/III.4.1/Tc grant under the TECHNETIUM and by the DEC-2/2021/IDUB/V.6/Si grant under the SILICIUM – ‘Excellence Initiative – Research University’ program is gratefully acknowledged. N. S. thanks the TASK Computational Centre and PLGrid Infrastructure for access to computational resources.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. R. Lawson and R. L. Melen, Organometallic Chemistry, The Royal Society Of Chemistry, 2017, vol. 41, pp. 1–27 Search PubMed.
- J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968–4971 CrossRef CAS PubMed.
- J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 RSC.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592–3612 RSC.
- P. Kolle and H. Nöth, Chem. Rev., 1985, 85, 399–418 CrossRef.
- W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016–5036 CrossRef CAS PubMed.
- H. Noeth, R. Staudigl and H. U. Wagner, Inorg. Chem., 1982, 21, 706–716 CrossRef CAS.
- P. Kölle and H. Nöth, Chem. Ber., 1986, 119, 313–324 CrossRef.
- Y. Shoji, N. Tanaka, K. Mikami, M. Uchiyama and T. Fukushima, Nat. Chem., 2014, 6, 498–503 CrossRef CAS PubMed.
- Y. Shoji, N. Tanaka, D. Hashizume and T. Fukushima, Chem. Commun., 2015, 51, 13342–13345 RSC.
- N. Tanaka, Y. Shoji, D. Hashizume, M. Sugimoto and T. Fukushima, Angew. Chem., Int. Ed., 2017, 56, 5312–5316 CrossRef CAS PubMed.
- S. Courtenay, J. Y. Mutus, R. W. Schurko and D. W. Stephan, Angew. Chem., Int. Ed., 2002, 41, 498–501 CrossRef CAS PubMed.
- C. J. Major, K. L. Bamford, Z.-W. Qu and D. W. Stephan, Chem. Commun., 2019, 55, 5155–5158 RSC.
- K. L. Bamford, Z.-W. Qu and D. W. Stephan, J. Am. Chem. Soc., 2019, 141, 6180–6184 CrossRef CAS PubMed.
- T. S. De Vries, A. Prokofjevs and E. Vedejs, Chem. Rev., 2012, 112, 4246–4282 CrossRef CAS PubMed.
- X. Tan and H. Wang, Chem. Soc. Rev., 2022, 51, 2583–2600 RSC.
- J. M. Farrell, J. A. Hatnean and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 15728–15731 CrossRef CAS PubMed.
- S. E. Denmark and Y. Ueki, Organometallics, 2013, 32, 6631–6634 CrossRef CAS PubMed.
- A. Prokofjevs, A. Boussonnière, L. Li, H. Bonin, E. Lacôte, D. P. Curran and E. Vedejs, J. Am. Chem. Soc., 2012, 134, 12281–12288 CrossRef CAS PubMed.
- T. S. De Vries, A. Prokofjevs, J. N. Harvey and E. Vedejs, J. Am. Chem. Soc., 2009, 131, 14679–14687 CrossRef CAS PubMed.
- B. Su, Y. Li, Z. H. Li, J.-L. Hou and H. Wang, Organometallics, 2020, 39, 4159–4163 CrossRef CAS.
- N. Szynkiewicz, A. Ordyszewska, J. Chojnacki and R. Grubba, RSC Adv., 2019, 9, 27749–27753 RSC.
- A. Ordyszewska, N. Szynkiewicz, E. Perzanowski, J. Chojnacki, A. Wiśniewska and R. Grubba, Dalton Trans., 2019, 48, 12482–12495 RSC.
- A. Ordyszewska, N. Szynkiewicz, J. Chojnacki and R. Grubba, Inorg. Chem., 2022, 61, 4361–4370 CrossRef CAS PubMed.
- N. Szynkiewicz, J. Chojnacki and R. Grubba, Inorg. Chem., 2020, 59, 6332–6337 CrossRef CAS PubMed.
- N. Szynkiewicz, A. Ordyszewska, J. Chojnacki and R. Grubba, Inorg. Chem., 2021, 60, 3794–3806 CrossRef CAS PubMed.
- I. Krossing, Chem. – Eur. J., 2001, 7, 490–502 CrossRef CAS PubMed.
- Calculated at the M06-2X//6-31+G(d,p) level of theory, including the presence of a solvent (CH2Cl2, CPCM Model) .
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- Calculated at the M06-2X//6-31+G(d,p) level of theory in the gas phase.
- D. Scheschkewitz, H. Amii, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Science, 2002, 295, 1880–1881 CrossRef CAS PubMed.
- T. Brückner, F. Fantuzzi, T. E. Stennett, I. Krummenacher, R. D. Dewhurst, B. Engels and H. Braunschweig, Angew. Chem., Int. Ed., 2021, 60, 13661–13665 CrossRef PubMed.
- C. Major, Z. W. Qu, S. Grimme and D. W. Stephan, Chem. – Eur. J., 2022, 28, e202200698 CrossRef CAS PubMed.
- E. N. Daley, C. M. Vogels, S. J. Geier, A. Decken, S. Doherty and S. A. Westcott, Angew. Chem., Int. Ed., 2015, 54, 2121–2125 CrossRef CAS PubMed.
- A. M. Borys, E. F. Rice, G. S. Nichol and M. J. Cowley, J. Am. Chem. Soc., 2021, 143, 14065–14070 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic, and computational details. CCDC 2163474–2163477 and 2184829. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc02933b |
| This journal is © The Royal Society of Chemistry 2022 |