
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective insertion of cyclopropenes into Mg–Mg bonds†
Feriel
Rekhroukh‡
a,
Linxing
Zhang‡
 ab,
Richard Y.
Kong
a,
Andrew J. P.
White
a and
Mark R.
Crimmin
*a
ab,
Richard Y.
Kong
a,
Andrew J. P.
White
a and
Mark R.
Crimmin
*a
aDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, 82 Wood Lane, Shepherds Bush, London, W12 0BZ, UK. E-mail: m.crimmin@imperial.ac.uk
bShenzhen Bay Laboratory, Shenzhen, 518132, P. R. China
First published on 28th June 2022
Abstract
The reaction of cyclopropenes with compounds containing Mg–Mg bonds is reported. 1,2-Dimagnesiation occurs exclusively by syn-addition to the least hindered face of the alkene forming a single diastereomeric product. DFT calculations support a concerted and stereoselective mechanism. These findings shed new light on the stereochemistry of reactions involving magnesium reagents.
The direct functionalisation of alkenes is an essential component of modern organic chemistry. Since the pioneering contribution of Markovnikov,1 it has been clear that controlling the selectivity of this reaction is of vital importance. While the regioselectivity of alkene functionalisation can be determined by assignment of the reaction products, for simple linear alkenes the stereoselectivity (syn- vs. anti-addition) is often a hidden consideration. In this regard, cyclic alkenes – specifically cyclopropenes – offer an opportunity to study the stereochemistry of alkene functionalisation.
Protocols for the hydroelementation and carboelementation of cyclopropenes have been reported. For example, a number of selective catalytic methods for the hydroboration,2,3 hydrostannation,4 and hydrosilylation5 of cyclopropenes have emerged in the recent years. Similarly, selective catalytic carbozincation6,7 and carbomagnesiation8 reactions of cyclopropenes have been achieved. These reactions typically employ organozinc or organomagnesium compounds in combination with a transition metal catalyst. In almost all cases, exclusive syn-addition to the cyclopropene occurs to give a single diastereomer, suggestive of a concerted and stereospecific transition state for carbometallation. Direct isolation and structural characterisation of the organometallic intermediates in these reactions is, however, uncommon; they are usually subjected to subsequent synthetic transformations without isolation.
Very recently the scope of main group reagents that can be used to functionalise alkenes has been expanded to compounds that contain reactive Mg–Mg bonds.9–11 Both Jones and coworkers,12–14 and our group,15 have demonstrated the 1,2-dimagnesiation of alkenes. In specific cases the reaction has been shown to be reversible. Despite the importance of these findings, the stereochemistry of addition remains unknown. In this paper, we report the 1,2-dimagnesiation of cyclopropenes. For the cases investigated, the reaction is stereoselective and occurs by syn-addition to the least hindered face of the cyclopropene. DFT calculations support a concerted reaction mechanism. We also investigate the stereointegrity of the resulting organomagnesium compounds. Despite the known propensity of stereocentres adjacent to Mg to undergo epimerisation,16–26 the syn-addition products are stable up to 60 °C.
Reaction of 1a with cyclopropene 2a, at 25 °C in C6D6 led to the rapid formation of the 1,2-dimagnesiocyclopropyl complex 3a. Similar reactivity was observed between 1a and 2b (15 min at 25 °C) or 2c (4 h at 60 °C), leading to the corresponding species 3b and 3c. The slowest reaction occurs with the most sterically hindered substrate. In all cases, the selective syn-insertion of the alkene into the Mg–Mg bond was observed. For 2a and 2b, attack of 1a occurs on the least hindered face of the ring system leading to the formation of a single diastereomer of the product (Scheme 1).
 | ||
| Scheme 1 Stereoselecive 1,2-dimagnesiation of cyclopropenes. | ||
Isolation and characterisation of 3a-c was possible by further reaction with either DMAP or THF to form 3a·(DMAP)2, 3b·(THF)2, and 3c·(THF)2. NMR spectroscopic data are consistent with symmetric structures. The most salient NMR data are the 1H and 13C signals observed for methine position directly linked to the Mg centre, which are observed in the following ranges: δH = –0.19 to –0.70 ppm and δC = 33.1 to 35.1 ppm. The structure of 3a·(DMAP)2 was confirmed by X-ray diffraction analysis (Fig. 1a). The extremely long Mg–Mg separation (>5.0 Å) along with the single bond C(1)–C(2) distance of 1.57(1) Å are consistent with the proposed insertion reaction. The Mg–C bonds of 2.02(1) and 2.20(1) Å are in the normal range for magnesium alkyls.12 Additional support for the proposed structure being retained in solution was obtained through in situ generation of an asymmetric analogue of 3a (with different ligands on each Mg centre). In this case, the two protons of the cyclopropane moiety become inequivalent. Analysis of the 1H NMR spectrum of the crude reaction mixture indicates a mutual 3JHH = 13.6 Hz coupling for the cyclopropane protons which is consistent with known literature values for syn-stereochemistry.27
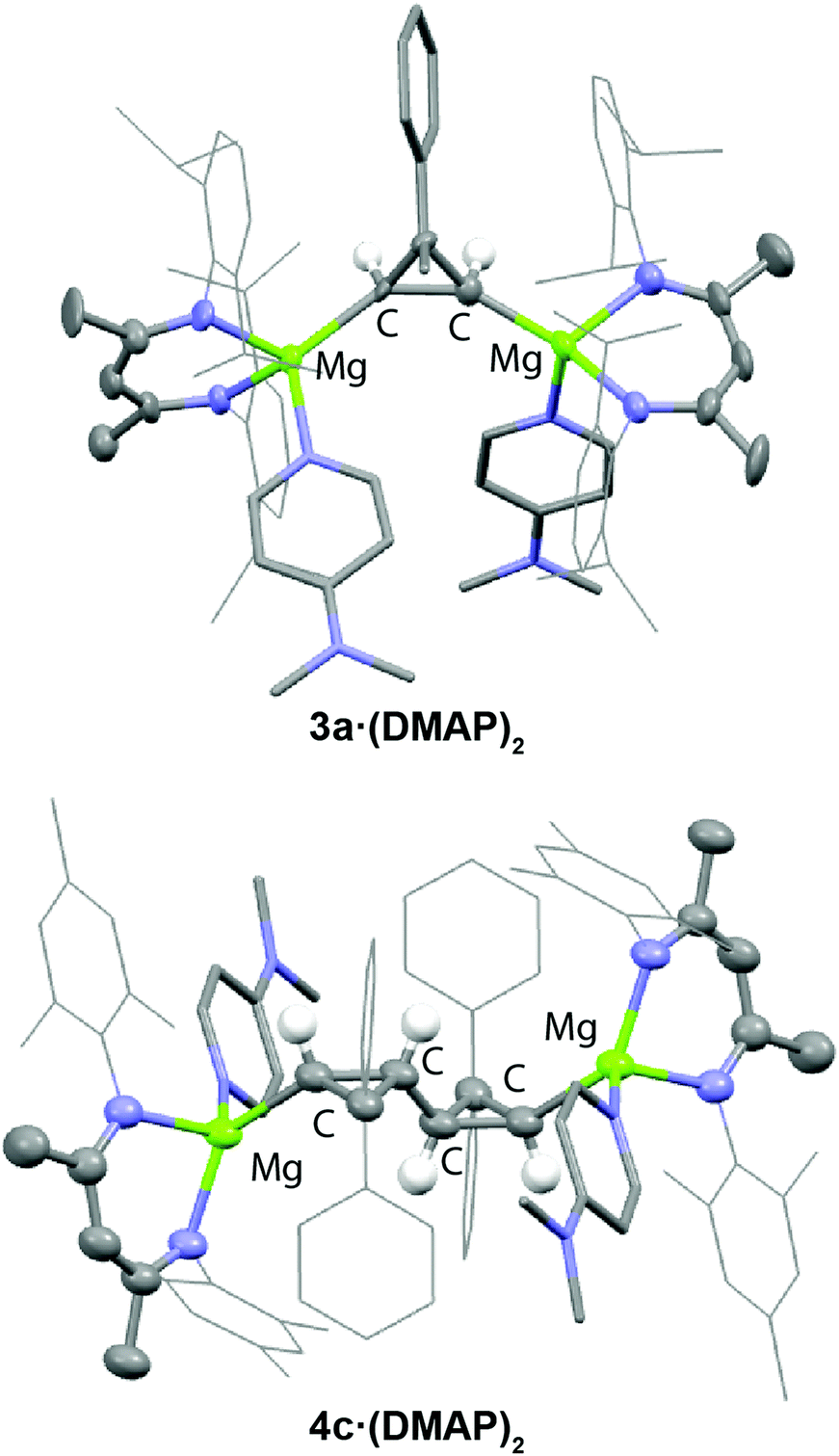 | ||
| Fig. 1 Structures of 3a·(DMAP)2 and 4c·(DMAP)2 from single crystal X-ray diffraction experiments. | ||
Addition of 2a-c (2 equiv.) to 1b, a less sterically hindered analogue of 1a, also proceeded extremely rapidly at 25 °C in C6D6 solution to form a single diastereomeric product. In these cases, however, a double insertion reaction occurred, forming 4a-c (Scheme 2). This reaction likely proceeds by two sequential steps: (i) a syn-selective insertion of the cyclopropene into the Mg–Mg bond of 1b and (ii) a syn-selective carbomagnesiation of a second equivalent of cyclopropene from the intermediate 1,2-dimagnesiocyclopropyl complex. This type of reactivity has already been reported by Jones and co-workers for reaction of 1b with ethylene in the presence of an N-heterocyclic carbene ligand.14 In the case of 2c, an intermediate 3d could be spectroscopically observed. Addition of a further equiv. of cyclopropane to this intermediate led to 4c.
 | ||
| Scheme 2 Sequential 1,2-dimagnesiation and carbometallation of cyclopropenes. | ||
The isolation of 4a-c was again achieved by reaction with DMAP. 4a-c·(DMAP)2 were isolated as yellow powders by precipitation in n-hexane in 50–75% yield. Spectroscopic data are consistent with their formulation. For example, for 4c·(DMAP)2, is characterised by two sets of doublets in the high field region of the 1H NMR spectrum at δ = 0.22 (2H, 3JHH = 10.5 Hz) and δ = 1.30 (2H, 3JHH = 10.5 Hz) ppm. The mutual coupling between these resonances was confirmed by a COSY NMR experiment. Clear evidence for the syn-selective double insertion is also evident from single-crystal X-ray diffraction of 4c·(DMAP)2 (Fig. 1b).
To gain a deeper understanding of the stereochemistry a series of plausible mechanisms were investigated by DFT calculations using the B3PW91 functional.28 Prior DFT studies have modelled the addition of ethylene to a derivative of 1b, but stereochemistry was not a consideration in this case.14 The reaction of 1b with cyclopropene 2a is calculated to proceed by an initial formation of a weak van der Waals complex Int-1 followed by a stereoselective syn-addition transition state TS-1 that connects directly to the product Int-2 (Fig. 2). Int-2 (R1 = Me, R2 = Ph) is a direct analogue of the spectroscopically observed intermediate 3d (R1 = R2 = Ph). This reaction is predicted to be exergonic by ΔG° (298 K) = –29.0 kcal mol−1 with a readily accessible activation barrier of ΔG‡ (298 K) = 5.7 kcal mol−1. An alternative syn-addition pathway in which 1b approaches the more hindered face of 2a was calculated to proceed by a slightly higher energy transition state TS-2 (ΔG‡ (298 K) = 9.3 kcal mol−1). The concerted anti-addition pathway, while leading to the most thermodynamically stable product, ΔG° (298 K) = –34.2 kcal mol−1, is prohibitively high in energy requiring distortion of the reactants into a twisted geometry in TS-3 (ΔG‡ (298 K) = 30.7 kcal mol−1). Hence, the calculations are consistent with the experiments and predict that the reaction of 1b with 2a should be under kinetic control and highly stereoselective at room temperature (ΔΔG‡ (298 K) = 3.6 kcal mol−1).
 | ||
| Fig. 2 (a) Free energy profile for mechanism of the first insertion. (b) Geometries of first insertion trasition states TS1-Me, TS1-Ph and TS1-anti. Computed at the PCM (benzene)/B3PW91-D3(BJ)/def2-TZVP//B3PW91/SDDAll-6-31G(d,p) level. | ||
Comparison of TS-1, TS-2, and TS-3 explains the origin of syn-selectivity. TS-1 is a four-membered transition state, and its structure determines the syn-addition to the least hindered face of the alkene. The geometry of TS-1 is asymmetric with one short Mg1–C1 (2.48 Å) and one long Mg2–C2 (3.08 Å) bond. TS-2 is similar. NBO calculations are also consistent with asynchronous bond formation occurring in TS-1, as evidenced by analysis of the NPA charges, which develop asymmetrically for both pairs of magnesium and carbon atoms (Fig. 3). The data indicate that the Mg1–C1 bond is formed earlier than the Mg2–C2 bond. In contrast, TS-3 is more symmetric, with almost no difference in two Mg–C bond distances (2.62 Å and 2.67 Å). This closer approach of the Mg–Mg reagent, combined with the required antarafacial bond formation to the π-system strongly disfavour the anti-addition by TS-3.
 | ||
| Fig. 3 NPA charges of the Me-facial syn-selective insertion Int-1, TS-1 and Int-2. Computed at the PCM (benzene)/B3PW91-D3(BJ)/def2-TZVP//B3PW91/SDDAll-6-31G(d,p) level. | ||
Further comparison of TS-1 and TS-2 can be used to rationalise the facial selectivity. QTAIM calculations combined with non-covalent interaction (NCI) analysis for reveals that TS-1 is stabilised by a greater number of (π-H–C) non-covalent interactions than TS-2 (Fig. 4). While we have not quantified these, it is likely a balance between these attractive dispersion interactions,29 and steric repulsion that favours addition to the face with the methyl substituent. The DFT model was expanded to consider chemoselectivity (see Fig. S30, ESI†). To simplify the possible reaction outcomes the substrate was modified to avoid the complication of facial selectivity. Calculations suggest that the syn-addition of two equiv. of the diphenyl substituted cyclopropene 2c to 1b to form the double insertion product 4c is energetically feasible. The first insertion occurs with a barrier of ΔG‡ (298 K) = 6.6 kcal mol−1, the second with a barrier of ΔG‡ (298 K) = 15.8 kcal mol−1, the overall reaction is exergonic by ΔG° (298 K) = –80.5 kcal mol−1. Hence, while the barrier to the second syn-insertion is higher than the first, both are expected to be accessible under the experimental conditions.
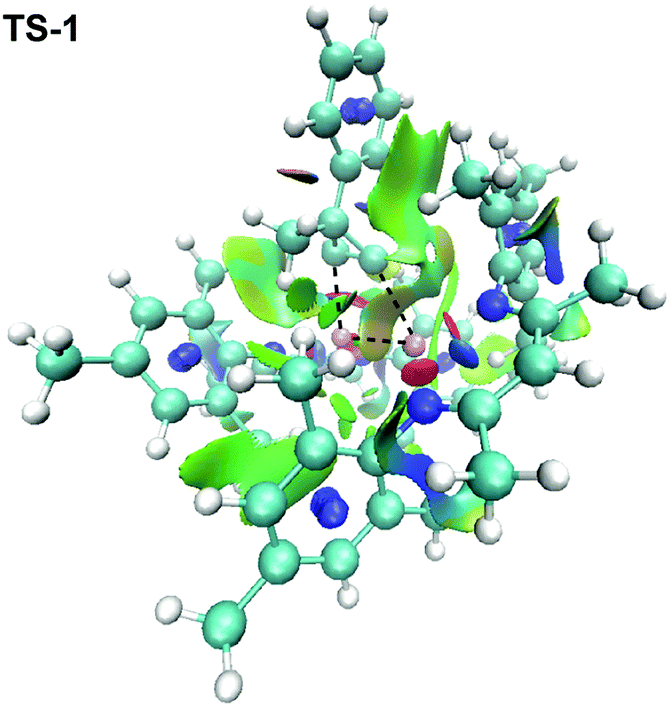 | ||
| Fig. 4 NCI plot of transition state TS-1. | ||
Isolation of 3b·(THF)2 - an organomagnesium compound with a defined stereochemistry – provides an opportunity to investigate the stereointegrity of centres adjacent to the magnesium site. Prior solution studies suggest that 2° alkyl magnesium reagents show increased configuration stability over 1° analogues.16–18,21,23,26 2° Alkyl magnesium reagents bearing cyclopropyl groups are expected to be the most stable derivatives, due to the low p-character of the α-carbon.19,20 For example, the half-life of racemisation of an enantiopure nitrile-substituted cyclopropyl magnesium reagent has been measured as 11 h at –100 °C22,24,25 in Et2O. 3b is formed under kinetic control due to the syn-addition pathway described above. The anti-isomer is expected to be thermodynamically more stable based on steric grounds. Monitoring 13.8 mM concentration solutions of 3b·(THF)2 in C6D6 solution over 120 h at 25 °C and then 120 h at 60 °C provided no evidence for epimerisation. The data suggest that 3b retains a syn-stereochemistry up to modest temperatures and that epimerisation in this system is not facile.
In summary, we have developed an experimental and computational understanding of the stereochemistry of 1,2-dimagnesiation of cyclopropenes. Key to this understanding is the realisation that the reaction occurs by a concerted syn-addition transition state in which the Mg–Mg bond adds across the most sterically accessible face of the alkene. These findings may have broad implications for the design of stereoselectivity reactions involving the addition of magnesium reagents to unsaturated carbon–carbon and carbon–heteroatom bonds.
We are grateful to the Leverhulme Trust for Funding (RPG 2020-006) and China Scholarship Council (CSC) for support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. Markownikoff, Justus Liebigs Ann. Chem., 1870, 153, 228–259 CrossRef.
- M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2003, 125, 7198–7199 CrossRef CAS PubMed.
- A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef CAS.
- M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2004, 126, 3688–3689 CrossRef CAS PubMed.
- L. Dian and I. Marek, Org. Lett., 2020, 22, 4914–4918 CrossRef CAS PubMed.
- M. Nakamura, A. Hirai and E. Nakamura, J. Am. Chem. Soc., 2000, 122, 978–979 CrossRef CAS.
- D. S. Müller and I. Marek, J. Am. Chem. Soc., 2015, 137, 15414–15417 CrossRef PubMed.
- Y. Cohen and I. Marek, Angew. Chem., Int. Ed., 2021, 60, 26368–26372 CrossRef CAS PubMed.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem. – Eur. J., 2010, 16, 938–955 CrossRef CAS PubMed.
- C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- A. J. Boutland, A. Carroll, C. Alvarez Lamsfus, A. Stasch, L. Maron and C. Jones, J. Am. Chem. Soc., 2017, 139, 18190–18193 CrossRef CAS PubMed.
- D. Dange, A. R. Gair, D. D. L. Jones, M. Juckel, S. Aldridge and C. Jones, Chem. Sci., 2019, 10, 3208–3216 RSC.
- K. Yuvaraj, I. Douair, L. Maron and C. Jones, Chem. – Eur. J., 2020, 26, 14665–14670 CrossRef CAS PubMed.
- R. Y. Kong and M. R. Crimmin, J. Am. Chem. Soc., 2020, 142, 11967–11971 CrossRef CAS PubMed.
- G. M. Whitesides and J. D. Roberts, J. Am. Chem. Soc., 1965, 87, 4878–4888 CrossRef CAS.
- G. M. Whitesides, M. Witanowski and J. D. Roberts, J. Am. Chem. Soc., 1965, 87, 2854–2862 CrossRef CAS.
- M. Witanowski and J. D. Roberts, J. Am. Chem. Soc., 1966, 88, 737–741 CrossRef CAS.
- H. M. Walborsky, F. J. Impastato and A. E. Young, J. Am. Chem. Soc., 1964, 86, 3283–3288 CrossRef CAS.
- H. M. Walborsky and A. E. Young, J. Am. Chem. Soc., 1964, 86, 3288–3296 CrossRef CAS.
- R. W. Hoffmann, Chem. Soc. Rev., 2003, 32, 225 RSC.
- P. R. Carlier and Y. Zhang, Org. Lett., 2007, 9, 1319–1322 CrossRef CAS PubMed.
- J. Beckmann and A. Schütrumpf, Org. Biomol. Chem., 2009, 7, 41–42 RSC.
- N. N. Patwardhan, M. Gao and P. R. Carlier, Chem. – Eur. J., 2011, 17, 12250–12253 CrossRef CAS PubMed.
- P. J. Rayner, P. O’Brien and R. A. J. Horan, J. Am. Chem. Soc., 2013, 135, 8071–8077 CrossRef CAS PubMed.
- S. Koller, J. Gatzka, K. M. Wong, P. J. Altmann, A. Pöthig and L. Hintermann, J. Org. Chem., 2018, 83, 15009–15028 CrossRef CAS PubMed.
- H. M. Hutton and T. Schaefer, Can. J. Chem., 1963, 41, 684–689 CrossRef CAS.
- M. J. Frisch, et al., Geometry optimizations were carried out using the B3PW91/SDDAll-6-31G(d,p). Single-point calculations were performed at SMD (benzene)/B3PW91-D3(BJ)/def2-TZVP-6-311+G(d,p) level. All calculations were conducted with Gaussian09, Gaussian, Inc., Wallingford CT, 2009. More details are included in the ESI.
- D. J. Liptrot, J. Guo, S. Nagase and P. P. Power, Angew. Chem., Int. Ed., 2016, 55, 14766–14769 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, details of calculations and characterization data (PDF). Coordinates for DFT calculations (xyz). Crystallographic data for 3a·(DMAP)2 and 4c·(DMAP)2 (cif). CCDC 2173920 and 2173921. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc02931f |
| ‡ Joint 1st authors. |
| This journal is © The Royal Society of Chemistry 2022 |