
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkyne insertion into Cu–Al bonds and selective functionalization to form copper acyl compounds†
Caitilín
McManus
 ,
Agamemnon E.
Crumpton
and
Simon
Aldridge
*
,
Agamemnon E.
Crumpton
and
Simon
Aldridge
*
Inorganic Chemistry Laboratory, Dept of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: simon.aldridge@chem.ox.ac.uk
First published on 24th June 2022
Abstract
We report on the insertion of alkynes into heterometallic M–M′ bonds, producing (aluminylalkenyl)copper compounds which possess differential reactivity at the two derived M–C functions. Uniquely, this system is capable of controlling access to isolable syn or anti dimetallated alkenes, by employing either kinetic or thermodynamic control. Subsequent derivatization with CO is both stereoselective (to syn systems) and regioselective (to Cu–C bonds), leading to the formation of the first structurally characterized examples of copper acyl compounds - aided by the cooperative reactivity of the proximal aluminium centre.
The 1,2-addition chemistry of alkynes is a key facet of their reactivity and is both widely exploited and well documented.1 In terms of organometallic compounds, both terminal and internal alkynes insert into M–E bonds (E = H, C, Hal),2–7 generating vinyl building blocks of considerable utility in organic synthesis. However, comparatively little is known about the insertion of alkynes into metal–metal bonds. Recent years have seen a limited number of reports of homo-dimetallation by direct insertion into M–M bonds, primarily for main group metals.8–10 Braunschweig's dialane system, for example, has been shown to effect the dialumination of 2-butyne (Fig. 1a).8 Isolated compounds formed by direct insertion of alkynes into hetero M–M′, or indeed M–E bonds (E = B, Si), are also relatively scarce.11 Pertinent to this work is the gold silyl system reported by Amgoune and Bourissou,12,13 which reacts under ‘drastic’ conditions with activated alkynes, via insertion into the Au–Si bond (with exclusively syn stereochemistry). Similarly, in 2021, Yamashita et al. reported an isomerization reaction via a retro-1,2-metal shift, in which the first step is syn insertion of an internal alkyne into a gold boryl complex (Fig. 1b).14
 | ||
| Fig. 1 (a) Insertion of 2-butyne into a dialane,8 (b) addition of Au–E bonds (E = B, Si) across C–C triple bonds.12–14 | ||
We recently reported the synthesis of a series of compounds containing direct aluminium-group 11 bonds, in which an aluminium centre supported by an NON ligand (NON = 4,5-bis(2,6-diisopropylanilido)-2,7-di-tert-butyl-9,9-dimethyl-xanthene) is bound to a group 11 phosphine fragment.15,16 Such compounds are among the small number of covalent aluminium-metal bonded species, and thus were perceived as prime candidates for insertion chemistry. Accordingly, it has been reported by ourselves and others that CO2 can insert into Al–M bonds, producing metallo-carboxylates.15–17 We now report the behaviour of the copper aluminyl complex 1 towards alkynes, which sequentially produces both syn and anti dimetallated alkenes bearing a copper and an aluminium group. Furthermore, we show that the differing reactivity of the metal substituents can be exploited, by selectively inserting CO to form isolable copper acyl compounds.
We chose to focus on studies of copper aluminyl complex (NON)AlCu(PtBu3), 1, in part because of the extensive use of organo-copper and -aluminium reagents in organic synthesis.18,19 Treatment of 1 with one equivalent of 3-hexyne (Scheme 1) leads to a rapid reaction giving a single species, characterized by a lone resonance in the 31P NMR spectrum at 55.5 ppm. The corresponding 1H spectrum shows two septets corresponding to the Dipp iPr methine protons, indicating a reduction in symmetry compared to the starting copper aluminyl compound, 1. Two new quartets arising from the CH2 protons of ethyl groups are found at 2.86 and 2.92 ppm; the signal at 2.86 ppm shows additional coupling (4JPH = 3 Hz) to the PtBu3 ligand, consistent with this ethyl group being situated geminal to the [CuPtBu3] moiety.
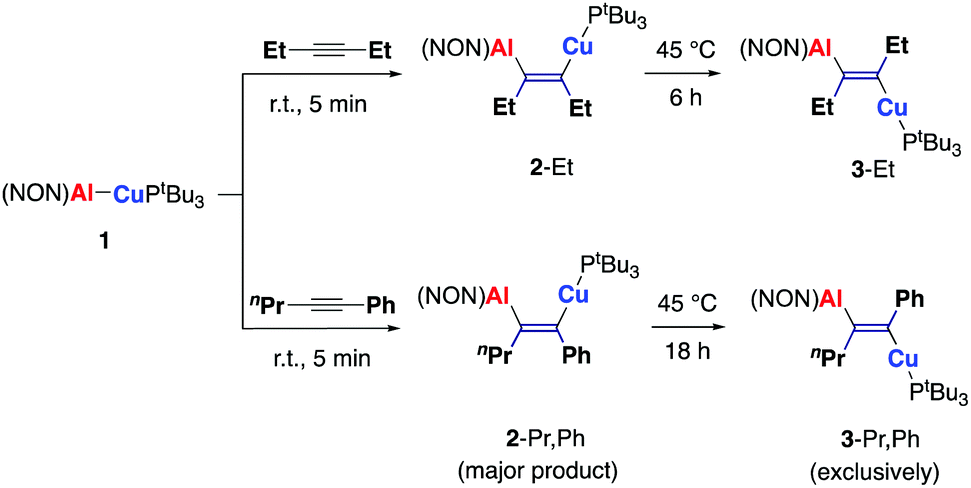 | ||
| Scheme 1 Reaction of copper aluminyl compound 1 with internal alkynes. | ||
Crystals suitable for X-ray crystallography obtained from a toluene solution allowed the solid-state structure of the product to be determined, confirming addition of the Al–Cu unit across the alkyne triple bond in syn fashion to give the aluminocuprated alkene, 2-Et (Fig. 2). To our knowledge, 2-Et represents the first example of an (aluminylalkenyl)copper complex. More broadly, while 1,2–dimetallated species have previously been synthesized via Pt-catalysed approaches,20 the formation of 2-Et represents the first structurally authenticated 1,2-hetero-dimetallation by insertion of an alkyne into an M–M′ bond.
 | ||
| Fig. 2 Molecular structures of 2-Et (left) an 3-Pr,Ph (right) in the solid state as determined by X-ray crystallography. Thermal ellipsoids set at the 50% probability level. Hydrogen atoms omitted and Dipp/tBu groups shown in wireframe for clarity. Key bond lengths (Å) and angles (°): (for 2-Et) C(49)–C(50) 1.352(4), Cu(1)–C(50) 1.940(2), Al(1)–C(49) 2.026(3), Cu(1)–P(1) 2.2096(7), C(60)–Cu(1)–P(1) 171.11(8), C(50)–C(49)–Al(1) 126.55(17), C(49)–C(50)–Cu(1) 127.93(19); (for 3-Pr,Ph) C(60)–C(61) 1.361(7), Al(1)–C(61) 1.974(4), Cu(1)–C(60) 1.919(4), Cu(1)–P(1) 2.2073(14), C(60)–Cu(1)–P(1) 178.89(17), C(60)–C(61)–Al(1) 119.6(4), C(61)–C(60)–Cu(1) 124.5(4). | ||
Although 2-Et can be cleanly isolated in crystalline form as the syn isomer, extended reaction times can be employed to enable onward reactivity, with complete consumption of 2-Et apparent after 24 h in toluene solution at room temperature. The 1H NMR spectrum at this point shows the formation of a second species, characterized by quartet and doublet of quartets signals for the alkenyl ethyl CH2 groups, similar to (but shifted from) those measured for 2-Et. The 31P NMR spectrum shows a new signal at δP = 58.5 ppm (cf. 55.5 ppm for 2-Et) and the corresponding 13C NMR spectrum displays a new downfield doublet at 183.0 ppm (2JPC = 71 Hz), corresponding to a [tBu3PCu]-bound alkenyl carbon (cf. δC = 188.3, 2JCP = 73 Hz for 2-Et). The spectroscopic evidence implies the formation of a closely related alumino-cuprated alkene, proposed to be the corresponding anti dimetallated alkene 3-Et (Scheme 1). While this assignment could not be confirmed crystallographically for 3-Et, comparison with the related (structurally characterised) system 3-Pr,Ph (see below) confirms that stepwise syn to anti isomerization is viable. As such, this system is uniquely capable of allowing selective access to isolable syn or anti dimetallated alkenes, by appropriate control of the reaction conditions.
The isomerisation reaction has features that are superficially similar to the chemistry of a boryl gold compound reported recently by Yamashita et al.14 In this system (depending on the alkyne), a transiently formed syn insertion product undergoes isomerisation to form either the anti disubstituted alkene, or a borylalkenylgold complex in which the R group has migrated to boron and the alkyne has assimilated a B-derived aryl function. The proposed intermediate in this reaction is a gold alkynyl-borate, which can produce both products via a 1,2-shift of either an R or Ar group. The operation of a similar mechanism in the conversion of 2-Et to 3-Et (Scheme 2) is supported by the fact that (NON)AlEt can be isolated from the reaction mixture as a minor product and structurally characterised (Fig. S36, ESI†). The formation of this product demonstrates unequivocally that C-to-Al migration of an Et group is viable, with the reverse step then providing a pathway for interconversion between the syn and anti isomers 2-Et and 3-Et. The formation of (NON)AlEt also implies concurrent generation of a copper acetylide co-product; while we were unable to isolate this compound, the lability of group 11 metal acetylides is well known, and the P-containing component was simply found to be ‘free’ tBu3P.
 | ||
| Scheme 2 Isomerisation of 2-Et to 3-Et; formation of (NON)AlEt as a side-product. | ||
To probe regioselectivity in the insertion of alkynes into 1, unsymmetrically substituted alkynes were also investigated (Scheme 1). The products of the reaction with 1-phenyl-1-butyne (PhCCEt) were not easily amenable to crystallization, so the closely related n-propyl alkyne, 1-phenyl-1-pentyne (PhCCnPr) was studied instead. The reactivity patterns of PhCCR (R = Et, nPr) towards 1 appear to be essentially identical.
Addition of PhCCnPr to a solution of 1 at 10 °C leads to the formation of a single product at very short reaction times (t < 20 min; Fig. S28, ESI†), but to a mixture of this compound and a second product in a ratio of ca. 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 after ca. 1 h, as determined by 31P NMR measurements (signals at δP = 60.2 and 61.5 ppm, respectively). After much longer reaction times this ratio is reversed (ca. 1:3 after 7 h). The 1H NMR spectrum of the first-formed product (2-Pr,Ph) features a resonance for the CH2CH2CH3 protons at 2.74 ppm, that shows no additional coupling to 31P. This signal is characteristic of an AA'XX' spin system, which is well known for n-propyl groups featuring restricted rotation.21 By contrast, the corresponding signal for the minor product (2-Ph,Pr) is a doublet of triplets, the result of additional 31P coupling to the CH2 group. These data imply that that the regiochemistry of the insertion process is such that the major initial product features the alkyl group geminal to the aluminium fragment (potentially on steric grounds), and Ph adjacent to the [CuPtBu3] unit. The minor product is the alternate regioisomer, in which the alkyl group is adjacent to [CuPtBu3]. Although the major product (2-Pr,Ph; Scheme 1) could not be crystallized, trapping experiments with CO (see below) confirm both the (syn) stereochemistry and the regio-chemistry of alkyne addition. Interestingly, a temporal plot shows that the concentration of 2-Pr,Ph reaches a maximum after ca. 60 min, and that the growth of the second-formed product 2-Ph,Pr then occurs at its expense (Fig. S28, ESI†). A potential explanation for this observation involves reversibility in the initial alkyne uptake step - a phenomenon which we could subsequently establish unequivocally by substitution of an inserted alkyne moiety by a different alkyne (see below).
:1 after ca. 1 h, as determined by 31P NMR measurements (signals at δP = 60.2 and 61.5 ppm, respectively). After much longer reaction times this ratio is reversed (ca. 1:3 after 7 h). The 1H NMR spectrum of the first-formed product (2-Pr,Ph) features a resonance for the CH2CH2CH3 protons at 2.74 ppm, that shows no additional coupling to 31P. This signal is characteristic of an AA'XX' spin system, which is well known for n-propyl groups featuring restricted rotation.21 By contrast, the corresponding signal for the minor product (2-Ph,Pr) is a doublet of triplets, the result of additional 31P coupling to the CH2 group. These data imply that that the regiochemistry of the insertion process is such that the major initial product features the alkyl group geminal to the aluminium fragment (potentially on steric grounds), and Ph adjacent to the [CuPtBu3] unit. The minor product is the alternate regioisomer, in which the alkyl group is adjacent to [CuPtBu3]. Although the major product (2-Pr,Ph; Scheme 1) could not be crystallized, trapping experiments with CO (see below) confirm both the (syn) stereochemistry and the regio-chemistry of alkyne addition. Interestingly, a temporal plot shows that the concentration of 2-Pr,Ph reaches a maximum after ca. 60 min, and that the growth of the second-formed product 2-Ph,Pr then occurs at its expense (Fig. S28, ESI†). A potential explanation for this observation involves reversibility in the initial alkyne uptake step - a phenomenon which we could subsequently establish unequivocally by substitution of an inserted alkyne moiety by a different alkyne (see below).
More forcing conditions (heating to 45 °C for 18 h) result in conversion of 2-Pr,Ph/2-Ph,Pr to a single species, characterized by a 31P resonance at 57.0 ppm. In this case, X-ray quality crystals of the product could be grown from hexane, and show a dimetallated alkene, 3-Pr,Ph in which the Al and Cu fragments are situated anti to one other, with the nPr group geminal to Al (Scheme 1 and Fig. 2). This regiochemical assignment is consistent with 2D NMR data for this compound in solution (Fig. S20, ESI†). The 1H NMR spectrum also bears a strong resemblance to that of 2-Pr,Ph, for example in featuring a resonance without 31P coupling (now at 3.11 ppm), arising from the CH2CH2CH3 protons. This observation provides further evidence that both 2-Pr,Ph and 3-Pr,Ph feature the same nPr/Ph regiochemistry. Moreover, the structural validation of the anti (aluminylalkenyl)copper fragment within the thermo-dynamic product 3-Pr,Ph supports the assignment of 3-Et as featuring the analogous anti alignment of the two ethyl fragments. As such, for both alkyne substrates, it is evident that the syn isomer is selectively formed initially (under kinetic control), and then reacts onwards to give exclusively the anti isomer under more forcing (thermodynamic) conditions (mild heating and longer reaction times), presumably due to the steric demands of the Al(NON) and CuPtBu3 fragments.
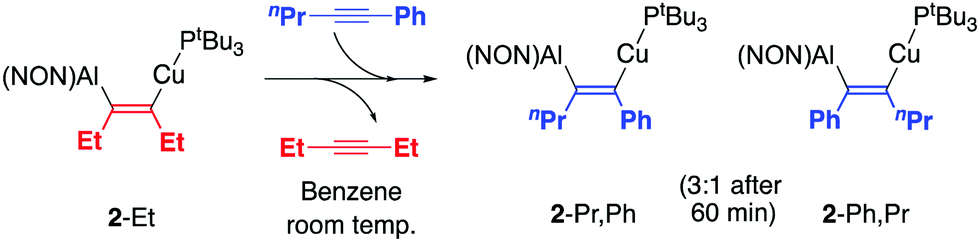 | ||
| Scheme 3 Exchange of the inserted alkyne fragment in 2-Et. | ||
Interestingly, exchange reactions show that the inserted alkyne fragment can be substituted for another, consistent with chemical reversibility in the insertion process. Accordingly, addition of one equivalent of PhCCnPr to a solution of 2-Et leads to the disappearance of the 31P NMR signal at 55.0 ppm associated with 2-Et, with accompanying growth of two signals associated with the major (60.2 ppm) and minor products (61.5 ppm) of PhCCnPr insertion (in the same ratio after 60 min as seen in the reaction of 1 itself with 1-phenyl-1-pentyne, Scheme 3). Consistently, one equivalent of free 3-hexyne is observed in the 1H NMR spectrum, showing that it has been released from 2-Et.
An alkene moiety substituted with two different metals provides a unique opportunity for onward functionalization chemistry, as was explored with β-silyl vinylgold(I) systems. Al-C and Cu–C bonds are known to possess differing hard/soft character, and therefore species such as 2-Et/2-Pr,Ph could be viewed as potentially discriminating bifunctional nucleophiles. In initial studies CO was chosen to explore onward functionalization, in part because such an insertion step might lead to the formation of an unprecedented copper acyl complex. Group 9 acyl compounds are well known, and critical in industrial hydroformylation.22,23 Likewise, Fe-bound acyl units can act as structural models of the Fe hydrogenase active site.24 For copper, however, these unstable species have been proposed as intermediates in Cu-catalysed reactions such as borocarbonylation and amidation,25 but have not been isolated, due to their propensity to undergo facile rearrangement.
Upon exposure of 2-Et to a CO atmosphere, an immediate colour change from colourless to yellow is seen, and quantitative conversion to a single new species observed in the 31P NMR spectrum (at 60.3 ppm). X-ray quality crystals grown from hexane allowed the structure of the product 4-Et to be determined (Fig. 3 and Scheme 4). This structure confirms that insertion of CO has occurred selectively into the Cu–C (rather than the Al–C) bond, forming a five-membered AlCCCO metallocycle featuring a pendant CuPtBu3 unit. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length (1.279(2) Å) is typical of an organic carbonyl function and the heterocycle is essentially planar. The 13C NMR spectrum of 4-Et shows a very low-field shifted doublet at 288.9 ppm, (2JCP = 66 Hz) arising from the copper-bound CO unit.26,27 The unusually high shift of the acyl group in 4-Et reflects O-coordination to the Lewis acidic Al centre, leading to deshielding of the carbonyl functionality. Consistent with this, the FT-IR spectrum of 4-Et displays a carbonyl stretching vibration at 1539 cm−1, i.e. significantly lower than typical CO stretches. The position and intensity of this band is in line with predictions made on the basis of DFT calculations (see ESI†).
O bond length (1.279(2) Å) is typical of an organic carbonyl function and the heterocycle is essentially planar. The 13C NMR spectrum of 4-Et shows a very low-field shifted doublet at 288.9 ppm, (2JCP = 66 Hz) arising from the copper-bound CO unit.26,27 The unusually high shift of the acyl group in 4-Et reflects O-coordination to the Lewis acidic Al centre, leading to deshielding of the carbonyl functionality. Consistent with this, the FT-IR spectrum of 4-Et displays a carbonyl stretching vibration at 1539 cm−1, i.e. significantly lower than typical CO stretches. The position and intensity of this band is in line with predictions made on the basis of DFT calculations (see ESI†).
 | ||
| Fig. 3 Molecular structures of 4-Et (left) and 4-Pr,Ph in the solid state as determined by X-ray crystallography. Thermal ellipsoids set at the 50% probability level. Hydrogen atoms omitted and Dipp/tBu groups shown in wireframe for clarity. Key bond lengths (Å) and angles (°): (for 4-Et) Cu(1)–C(2) 1.9255(16), Al(1)–O(1) 1.8843(12), Al(1)–C(5) 2.0027(17), O(1)–C(1) 1.279(2), C(5)–C(2) 1.362(2); (for 4-Pr,Ph) C(3)–C(2) 1.3597(19), O(1)–C(1) 1.2763(16), C(1)–C(2) 1.4908(18), Al(1)–O(1) 1.8806(10). | ||
 | ||
| Scheme 4 Selective insertion of CO into the Cu–C bond of 2-Et and 2-Pr,Ph. | ||
To our knowledge, 4-Et is the first example of a crystallographically characterised copper acyl complex. The proximity of the Lewis acidic aluminium site in 4-Et is implicit in this, offering additional stabilization of the acyl unit in the crystal via an O→Al interaction (d(O–Al) = 1.8843(12) Å) which completes a trigonal bipyramidal coordination geometry at aluminium (τ = 0.61). The presence of the proximal aluminium centre is also important mechanistically - with the CO insertion process being found to be exclusively accessible to systems featuring a syn configuration of the two metal centres. Presumably this reflects the fact that a short-lived copper carbonyl intermediate would be activated towards migration of the alkenyl fragment by Lewis acid activation at O. As such, the anti (aluminylalkenyl)copper system 3-Pr,Ph shows no reactivity towards CO under the conditions examined.
Utilizing this fact, the CO insertion reaction could also be exploited to trap the rapidly formed syn product 2-Pr,Ph generated in the reaction of 1 with PhCCnPr. Addition of CO to in situ generated 2-Pr,Ph results in the formation of the analogous five-membered metallocycle 4-Pr,Ph (Fig. 3). Moreover, this derivatization corroborates NMR evidence that the product formed by insertion of 1-phenyl-1-pentyne into 1 is 2-Pr,Ph, featuring the alkyl and aluminium fragments bound to the same alkenic carbon centre.
In conclusion, we report alkyne insertion into a heterometallic M–M′ bond, producing (aluminylalkenyl)copper compounds which possess differential reactivity at the two derived M–C functions. This system allows control over the nature of the product formed: kinetic control gives access to syn isomers with high selectivity, while the corresponding anti products can be obtained under more forcing conditions. Moreover, the use of short reaction times allows near-exclusive regio-control to be exerted over the insertion of mixed aryl/alkyl substituted alkynes. Longer reaction times generate mixtures of regio-isomers, with independent experiments showing that the initial insertion process is chemically reversible. One alkyne substrate is capable of displacing another. Stereochemically syn systems can be selectively derivatized by reaction with CO, leading to the formation of copper acyl compounds - aided by the cooperative reactivity of the proximal aluminium centre. Finally, metallocycles such as 4-Et can be hydrolysed quantitatively to free the organic fragment, with the α,β unsaturated aldehyde product (in this case (E)-2-ethylpent-2-enal) being formed via the net hydroformylation of an alkyne through the controlled action of an unsymmetrical bimetallic reagent.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. March and M. B. Smith, March's Advanced Organic Chemistry, John Wiley & Sons, Ltd, New Jersey, 2007, pp. 999–1014 Search PubMed.
- V. P. Ananikov and I. P. Beletskaya, Top. Organomet. Chem., 2013, 43, 1 CrossRef CAS.
- D. S. Müller and I. Marek, Chem. Soc. Rev., 2016, 45, 4552 RSC.
- D. Bag, S. Mahajan and S. D. Sawant, Adv. Synth. Catal., 2020, 362, 3948 CrossRef CAS.
- X. Li, S. He and Q. Song, Org. Lett., 2021, 23, 2994 CrossRef CAS PubMed.
- A. M. Suess, M. R. Uehling, W. Kaminsky and G. Lalic, J. Am. Chem. Soc., 2015, 137, 7747 CrossRef CAS PubMed.
- Y. Nishimoto and M. Yasuda, Chem. – Asian J., 2020, 15, 636 CrossRef CAS PubMed.
- A. Hofmann, A. Lamprecht, O. F. González-Belman, R. D. Dewhurst, J. O. C. Jiménez-Halla, S. Kachel and H. Braunschweig, Chem. Commun., 2018, 54, 1639 RSC.
- D. Dange, A. R. Gair, D. D. L. Jones, M. Juckel, S. Aldridge and C. Jones, Chem. Sci., 2019, 10, 3208 RSC.
- Y. Zhao, Y. Liu, Y. Lei, B. Wu and X. J. Yang, Chem. Commun., 2013, 49, 4546 RSC.
- For metal-catalysed diboration, see: E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef CAS PubMed.
- M. Joost, P. Gualco, S. Mallet-Ladeira, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2013, 52, 7160 CrossRef CAS PubMed.
- M. Joost, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgou-ne and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 10373 CrossRef CAS PubMed.
- (a) A. Suzuki, L. Wu, Z. Lin and M. Yamashita, Angew. Chem., Int. Ed., 2021, 60, 21007 CrossRef CAS PubMed For earlier examples of direct insertion of alkynes into a M–B bond, see for example:; (b) G. R. Clark, G. J. Irvine, W. R. Roper and L. J. Wright, Organometallics, 1997, 16, 5499 CrossRef CAS.
- C. McManus, J. Hicks, X. Cui, L. Zhao, G. Frenking, J. M. Goicoechea and S. Aldridge, Chem. Sci., 2021, 12, 13458 RSC.
- J. Hicks, A. Mansikkamäki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237 CrossRef CAS PubMed.
- (a) H.-Y. Liu, R. J. Schwamm, M. S. Hill, M. F. Mahon, C. L. McMullin and N. A. Rajabi, Angew. Chem., Int. Ed., 2021, 60, 14390 CrossRef CAS PubMed; (b) H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon and C. L. McMullin, Dalton Trans., 2022, 51, 3913 RSC.
- N. Yoshikai and E. Nakamura, Chem. Rev., 2011, 112, 2339 CrossRef PubMed.
- B. Breit and Y. Schmidt, Chem. Rev., 2008, 108, 2928 CrossRef CAS PubMed.
- I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320 CrossRef CAS PubMed.
- P. J. Stevenson, Org. Biomol. Chem., 2011, 9, 2078 RSC.
- T. Zhou, S. Malakar, S. L. Webb, K. Krogh-Jespersen and A. S. Goldman, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 3419 CrossRef CAS PubMed.
- R. R. Reinig, E. L. Fought, A. Ellern, T. L. Windus and A. D. Sadow, Dalton Trans., 2018, 47, 12147 RSC.
- B. Hu, D. Chen and X. Hu, Chem. – Eur. J., 2014, 9, 2078 Search PubMed.
- For copper acyl transient intermediates in Cu-catalysed reactions, see for example: (a) F. Wu, J. Holz, Y. Yuan and X. Wu, CCS Chem., 2020, 2, 2643 Search PubMed; (b) Y. Yuan, F. Wu, J. Xu and X. Wu, Angew. Chem., Int. Ed., 2020, 59, 17055 CrossRef CAS PubMed; (c) Q. Luo, C. Wang, W.-X. Zhang and Z. Xi, Chem. Commun., 2008, 1593 RSC; (d) H.-Q. Geng, T. Meyer, R. Franke and X.-F. Wu, Chem. Sci., 2021, 12, 14337 RSC.
- J. G. Cordaro and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 3432 CrossRef CAS PubMed.
- A. Haynes, P. M. Maitlis, G. E. Morris, G. J. Sunley, H. Adams, P. W. Badger, C. M. Bowers, D. B. Cook, P. I. P. Elliott, T. Ghaffar, H. Green, T. R. Griffin, M. Payne, J. M. Pearson, M. J. Taylor, P. W. Vickers and R. J. Watt, J. Am. Chem. Soc., 2004, 126, 2847 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray CIF. CCDC 2157971–2157975. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc02578g |
| This journal is © The Royal Society of Chemistry 2022 |