
A redox-switchable catalyst with an ‘unplugged’ redox tag†
Cristian L.
Gutiérrez-Peña
 ,
Macarena
Poyatos
* and
Eduardo
Peris
*
,
Macarena
Poyatos
* and
Eduardo
Peris
*
Institute of Advanced Materials (INAM), Centro de Innovación en Química Avanzada (ORFEO-CINQA). Universitat Jaume I, Av. Vicente Sos Baynat s/n, 12071-Castellón, Spain. E-mail: poyatosd@uji.es; eperis@uji.es
First published on 24th August 2022
Abstract
Two bis-(propyl-imidazolium)-napthalenediimide (NDI) salts were prepared and used as N-heterocyclic carbene (NHC) precursors for the preparation of dimetallic complexes of rhodium and iridium. Infrared spectroelectrochemical studies indicate that the metals are sensitive to changes in the electronic state of the NDI moiety. The catalytic behavior of the rhodium and iridium complexes was tested in the cycloisomerization of alkynoic acids, where the complexes showed effective redox-switching properties.
Redox-active ligands1 form part of the broader class of multifunctional ligands,2 a group that has experienced growing attention in the last few decades, mostly due to their applications in catalysis. Redox-active ligands offer advantages over synthetically-based approaches for altering the reactivity of catalysts. In particular, ligands bearing redox-active groups can control the electron-richness of the metal through ligand-centered oxidation or reduction processes without the need of further synthetic steps, and this is why redox-switchable catalysis is regarded as an atom-economical strategy for generating multiple species from a single precursor. It is widely accepted that the through-bond interaction is one of the main factors that determine the efficiency of the electronic communication between the metal and the redox active unit, so one of the basic rules for designing a redox-switchable ligand is that the redox-active center must remain close to the metal and preferably connected through conjugated bonds.3 N-Heterocyclic carbenes (NHCs) have been particularly fruitful in the design of redox-switchable ligands,3,4 mostly because their preparation is relatively simple, and because they can be appended to essentially any functional group.2a We recently described two NHC ligands fused to naphtalenediimide (NDI) and perylene-diimide (PDI) moieties that were coordinated to rhodium, iridium5 and gold.6 The presence of the NDI or PDI units appended to the NHC ligand allowed toggling between three levels of electronic control of the metal, and this was used for studying the redox-switchable catalytic properties of the complexes in the cycloisomerization of alkynoic acids,5a and in the hydroamination of alkynes.6 In view of these previous results, we herein describe the preparation of a series of dimetallic rhodium and iridium complexes in which the two metals are bound by a NDI-di-propylene-di-imidazolylidene ligand. Given that, apparently, the redox-active NDI unit is electronically disconnected from the two imidazolylidene groups due to the presence of the two n-propyl chains, it is expected that the modification of the oxidation state of the NDI core should impart little or negligible influence on the electron-donating ability of the two NHC units. We show that, contrary to what it is expected, the reduction of the NDI fragment has a strong influence on the electron richness of the metal, and on the redox-switchable catalytic properties of the resulting metal complexes.
For the preparation of our NDI-functionalized di-NHCs, we used the N,N’-bis(3-propylimidazolium)-1,4,5,8-naphtalenediimide salts [1A](Br)2 and [1B](Br)2.7 The preparation of the rhodium and iridium complexes 2A/B and 3A/B was performed by transmetallation of the NHC ligand from the in situ preformed silver–NHC complexes, following the procedure depicted in Scheme 1. The resulting complexes were isolated as yellow solids with yields ranging from 81–95%, and were characterized by NMR spectroscopy and Electrospray Ionization Mass Spectrometry (ESI-MS). The 13C NMR spectra of the rhodium complexes 2A and 2B show the distinctive resonances due to the metallated carbene carbons as doublets (1JRh–C = 51.0 Hz) at 182.1 and 183.3 ppm, respectively. The 13C NMR resonances due to the metallated carbene carbons of the iridium complexes 3A and 3B are observed at 180.1 and 180.9 ppm, respectively.
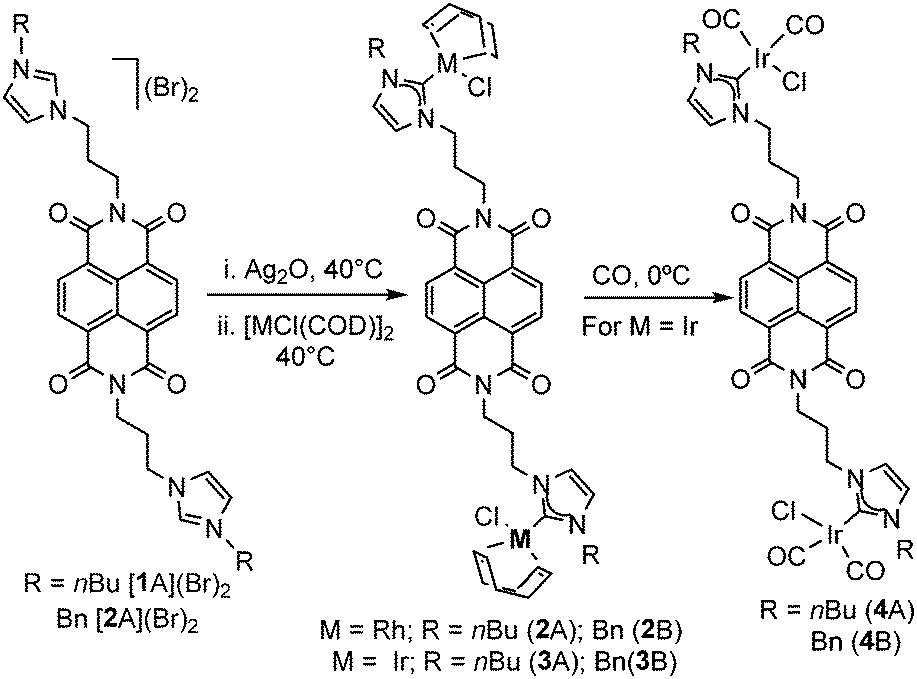 | ||
| Scheme 1 Preparation of NDI-functionalized rhodium and iridium di-NHC complexes. | ||
The molecular structure of the iridium complex 3B was unambiguously confirmed by single crystal X-ray diffraction studies (Fig. 1). The structure consists of two iridium-chloro-(1,5-cyclooctadiene) moieties bound by a naphthalene-di-isopropyl-connected di-NHC ligand, and each of the NHC rings is functionalized with a N-benzyl group. The distance of the Ir–Ccarbene bond is 2.044(7) Å. All other distances and angles are within the expected range.
 | ||
| Fig. 1 Molecular structure of 3B. Hydrogen atoms and solvent (CHCl3) are omitted for clarity. The N-benzyl groups are represented in the wireframe mode for clarity. | ||
In order to obtain information about the electrondonating character of the di-NHC ligands, the iridium carbonyl complexes 4A and 4B were obtained by bubbling CO in CH2Cl2 solutions of 3A and 3B (Scheme 1). The IR (CH2Cl2) spectra of these complexes show two bands due to the stretching of the C–O bonds at 2080 and 2000 cm−1 (for 4A), and 2081 and 1999 (for 4B).
| Compound | E 21/2 (V)/ΔEb (mV) | E 11/2 (V)/ΔE (mV) | E pa (V) |
|---|---|---|---|
| a Measurements performed at a scan rate of 100 mV s−1 and referenced vs. ferrocenium/ferrocene. b The second reduction event is irreversible for complexes 2, 3 and 4, the value indicated in the table corresponds to the cathodic peak potential (Epc). c The oxidation event is irreversible for complexes 2 and 3, the value shown in the table corresponds to the anodic peak potential (Epa). | |||
| [1A](Br)2 | −1.30/71 | −0.92/75 | — |
| [1B](Br)2 | −1.29/71 | −0.92/91 | — |
| 2A | −1.53 | −1.06/91 | 0.39 |
| 2B | −1.47 | −1.03/77 | 0.45 |
| 3A | −1.44 | −1.03/77 | 0.44 |
| 3B | −1.49 | −1.02/79 | 0.43 |
| 4A | −1.42 | −1.00/78 | — |
| 4B | −1.41 | −0.97/81 | — |
The cyclic voltammetry (CV) studies of compounds 1–4(Table 1) reveal two well-separated redox events associated to the sequential one-electron reduction of the NDI core. For the iridium and rhodium complexes 2–4, the cyclic voltammograms show one reversible reduction wave followed by a second irreversible process. The reduction potentials of the iridium and rhodium complexes 2–4 are slightly more negative (∼100 mV) than those shown by the salts [1A](Br)2 and [1B](Br)2, as consequence of the neutral character of the metal complexes compared to the dicationic nature of the NDI-functionalized bis-imidazoliums. The values of the reduction potentials of the NDI core of all four metal complexes (2–4) are virtually identical, indicating that the reduction of the NDI core is not sensitive to the change of the metal, nor to changes in the metal coordination sphere. In the case of the COD iridium and rhodium complexes 2 and 3, an irreversible oxidation wave is also observed.
For studying the nature and the stability of the species formed upon reduction of the NDI moiety, we performed the UV-Vis spectroelectrochemical (SEC) experiments of [1A](Br)2 and 3A, which we chose as model compounds (see ESI† for details). The UV-Vis spectrum of [1A]2+, shows an intense vibronically resolved band centered at 350 nm. Upon application of stepwise reduction potential, this band disappears and a new intense band at 475 nm associated to [1A˙]+ appears. Further reduction of this species gives rise to the appearance of two vibronically resolved new bands at 425 and 600 nm, which are associated to the doubly-reduced species 1A. The UV-Vis SEC experiments performed using the 3A allowed observing the transition between the neutral complex 3A (vibronically-coupled band at 375 nm), [3A]− (band at 490 nm), and [3A]2− (bands at 400, 475 and 590 nm), as the reduction potential was gradually increased (see ESI† for details). In addition, once 2-electron-reduced species 1A and [3A]2− were formed, the application of positive potentials allowed recovering [1A]2+ and 3A, respectively, thus indicating the reversibility of the process.
Then, we sought to perform infrared SEC (IR-SEC) experiments on the iridium carbonyl complex 4A. As can be observed on the series of infrared difference spectra shown in Fig. 2, the progressive reduction of 4A is accompanied by the disappearance of the two bands at 2002 and 2080 cm−1, and the appearance of two new bands at 1993 and 2070 cm−1, which are assigned to the one-electron reduced compound [4A˙]−. Further reduction to lower potentials results in the disappearance of these two bands with the concomitant appearance of two new C–O stretching bands at 1989 and 2068 cm−1, assigned to the 2e-reduced compound [4A]2−. These results indicate that the one-electron reduction of the NDI unit results in an average ΔCO shift of −9.5 cm−1, while the second reduction produces a further shift of −3 cm−1, thus indicating that the reduction of the NDI unit produces an important increase of the electron-donating character of the NDI-di-NHC ligand. This result is important for two reasons. First, we need to consider that the effect of the reduction of the NDI core needs to be splitted between the two NHCs of the ligand, and therefore the ‘per-NHC’ effect should be expected to be lower than if the NDI unit was connected to a single NHC unit. And second, the fact that the electronic connection between the NDI moiety and the NHCs is disrupted by the n-propyl chains throws into question the need of considering the distance between the redox unit and the metal, as well as their good electronic communication, as critical parameters to be considered for building effective redox-switchable ligands. Since we are aware that these results may seem rather counter-intuitive, we performed two experiments in order to confirm the increase of the electron-donating character of the NDI-functionalized di-NHC ligand upon reduction of the NDI core. First, we performed an IR-SEC experiment of a dichloromethane solution containing an equimolar amount of [IrCl(IBu)(CO)2] (IBu = 1,3-di-n-butyl-imidazolylidene) and N,N’-di-nbutyl-naphthalene-diimide. Upon progressive reduction of the NDI molecule, we did not observe any changes in the frequencies of the C–O stretching bands of the iridium-carbonyl complex (see Fig. S38 in ESI†). This experiment indicates that the reduction of the NDI moiety has an influence on the electronic nature of the metal only if the NDI is tethered to the binding unit of the ligand, and discards the possibility that an ‘outer-sphere’ or a direct binding interaction between the NDI molecule and the metal may be responsible of the effect. Second, we obtained the tungsten complex [NDI-(NHC)2][W(CO)5]2 (see ESI† for details). Determination of changes in the carbonyl stretching frequencies of group 6 [M(CO)5] can also be used for assessing changes in the electron-donating power of redox-switchable ligands.3 The IR-SEC experiment on a CH2Cl2 solution of [NDI-(NHC)2][W(CO)5]2 showed that the reduction of the NDI moiety decreased the νA1(ax) frequency in 91 cm−1, in great accordance with previously results reported by other authors.8 The bands due to the equatorial carbonyl groups also experience a significant decrease of the frequency (νA1(eq) = 6 cm−1, νE(eq) = 16 cm−1, as can be observed in Fig. S39, ESI†).
 | ||
| Fig. 2 Infrared difference spectra resulting from the IR-SEC reduction of 4A in CH2Cl2 (0.25 M [N(nBu)4][PF6]). The electrochemical reduction was performed applying progressively lower potentials with a Au working electrode, Pt counter electrode, and Ag wire pseudo-reference. | ||
We decided to explore the catalytic properties of 2A and 3A in the cycloisomerization of alkynoic acids. This reaction can be catalysed by rhodium and iridium complexes, as has been studied by some authors,9 including us.10 We recently showed that a series of NDI-functionalized NHC complexes of iridium and rhodium could be used effectively as redox-switchable catalysts in the cycloisomerization of alkynoic acids,5a and found that the rate determining step of the catalytic cycle involves the oxidative addition of the alkynoic acid to the metal.
We first studied the cycloisomerization of 4-pentynoic acid. The reactions were carried out in acetonitrile at 80 °C, using a catalyst loading of 0.1 mol% (based on the amount of metal). The resulting time-dependent reaction profiles are shown in Fig. 3. As can be seen from the plots, the activity of the rhodium and iridium catalysts are virtually identical, as reflected by the very similar rate constants (2.3 × 10−6 Ms−1 for 2A and for 2.1 × 10−6 Ms−1 for 3A). Under these reaction conditions, both catalysts afford full conversion after 48 hours of reaction. By studying the time-dependent reaction profiles at different concentrations of catalyst 2A, we determined that the reaction is first order in catalyst (see ESI† for details). Given that cobaltocene has a redox potential of −1.33V,11 it constitutes a suitable agent for the selective one-electron reduction of 2A and 3A. As can be observed in Fig. 3, the addition of cobaltocene (one equivalent with respect to the catalyst) produced a twofold increase in the kinetic constant of the reaction catalysed by the rhodium complex 2A (from 2.3 × 10−6 Ms−1 to 4.3 × 10−6 Ms−1). This enhancement is even more pronounced in the case of the reaction catalysed with the iridium complex 3A, for which addition of cobaltocene produces an increase of more than one order of magnitude (from 2.1 × 10−6 Ms−1 to 3.1 × 10−5 Ms−1). These observations are in agreement with our previous results obtained for the rhodium and iridium catalysts obtained with an NDI moiety fused to the NHC ligand,5a although in the present case the enhancement of the catalytic activity upon reduction of the NDI moiety is slightly lower, as one would expect for the lower increase of the per-NHC electron-donating character, as we pointed out above.
 | ||
| Fig. 3 Time-dependent reaction profiles for the cyclization of 4-pentynoic acid using catalysts 2A and 3A with and without addition of cobaltocene. The reactions were carried out in acetonitrile, with an initial concentration of 4-pentynoic acid of 0.33 M, and a catalyst loading of 0.1 mol%. Cobaltocene was added in a 0.1 mol% with respect to the substrate. Yields were determined by 1H-NMR spectroscopy, using 1,3,5-trimethoxybenzene as integration standard. The figure also shows the kinetic constants for each reaction. | ||
We also evaluated if we could toggle between the active and inactive forms of the iridium catalyst 3A along the course of the reaction (Fig. 4). We first allowed the reaction to evolve for 2 hours in the presence of 0.1 mol% of 3A, and the reaction produced 8% of the lactone product. Then, cobaltocene was added, and the reaction was let to evolve for one more hour, after which 74% of product was formed. Finally, acetylferrocenium tetrafluoroborate ([Fc’](BF4)) was added, and the reaction slowed down to equal the rate of the initial period when the reaction was performed in the absence of cobaltocene (see Fig. 4 for comparing the rate constants). These results show how the catalyst can be switched on by the addition of a reducing agent, and the switched back off by further adding an oxidant.
 | ||
| Fig. 4 Plot showing the cycloisomerization of 4-pentynoic acid with 0.1 mol% of 3A and sequential additions of [CoCp2] and acetyl ferrocenium tetrafluoroborate, [Fc’](BF4) (blue solid dots). The plot showing the evolution of the reaction using 3A + [CoCp2] (both 0.1 mol%) is also shown for comparison (empty red dots). The reactions were carried out in CH3CN at 80 °C, with an initial concentration of 4-pentynoic acid of 0.33M. Lines are used to guide the eye. | ||
We also performed the reaction with the more challenging 5-pentynoic acid using 3A, with a catalyst loading of 1 mol%, in acetonitrile at 80 °C. For this reaction we did not observe the formation of product after 24 hours. When we carried out the reaction adding 1 mol% of cobaltocene, then we observed that the product yield raised to 82% after seven hours (see ESI† for reaction profiles).
Finally, given the doubly switchable character of the NDI-functionalized ligand, we sought to study the catalytic performance of 3A in the presence of a stronger reductant such as decamethylcobaltocene ([CoCp*2]), which facilitates the 2e-reduction of the NDI moiety. Under these conditions, the cycloisomerization of 4-pentynoic and 5-alkynoic acids are significantly improved with respect to the results obtained in the absence of this strong reductant (see Table S5 in ESI†). However, the outcome of the reactions is worse than when performed in the presence of [CoCp2]. As we reported previously,5a the addition of a reductant accelerates the catalytic process, because it facilitates the oxidation of the catalyst at the oxidative addition step of the cycle, which is the rate-determining step of the cycle. However, if a stronger reductant is added (such as [CoCp*2]), then the final reductive elimination step of the cycle can be hampered, and this can be detrimental for the overall catalytic process.
In summary, our studies indicate that the redox-switchable character of our catalyst is maintained, despite showing an ‘apparently’ disrupted electronic communication between the NDI tag and the metal centre. Although further detailed studies need to be carried out, our results seem to indicate that for an effective control of the redox-switchable character of the catalyst strong electronic communication between the redox tag and the metal may not be always required.
We gratefully acknowledge financial support from the Ministerio de Ciencia y Universidades (PGC2018-093382-B-I00) and the Universitat Jaume I (UJI-B2020-01 and UJI-B2021-39). We are grateful to the Serveis Centrals d’Instrumentació Científica (SCIC-UJI) for providing with spectroscopic facilities. We are also grateful to the Santiago Grisolía Program (C.L. G.-P.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. M. Allgeier and C. A. Mirkin, Angew. Chem., Int. Ed., 1998, 37, 894–908 CrossRef; (b) L. A. Berben, B. de Bruin and A. F. Heyduk, Chem. Commun., 2015, 51, 1553–1554 RSC; (c) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS; (d) B. de Bruin, Eur. J. Inorg. Chem., 2012, 340–342 Search PubMed; (e) O. R. Luca, D. L. Huang, M. K. Takase and R. H. Crabtree, New J. Chem., 2013, 37, 3402–3405 RSC; (f) P. J. Chirik, Inorg. Chem., 2011, 50, 9737–9740 CrossRef CAS PubMed; (g) A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969–1992 CrossRef CAS PubMed; (h) J. Wei and P. L. Diaconescu, Acc. Chem. Res., 2019, 52, 415–424 CrossRef CAS PubMed.
- (a) E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed; (b) A. D. Burrows, Sci. Prog., 2002, 85, 199–217 CrossRef CAS PubMed; (c) R. H. Crabtree, New J. Chem., 2011, 35, 18–23 RSC; (d) W. I. Dzik, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Angew. Chem., Int. Ed., 2011, 50, 3356–3358 CrossRef CAS PubMed; (e) H. Griitzmacher, Angew. Chem., Int. Ed., 2008, 47, 1814–1818 CrossRef PubMed.
- E. L. Rosen, C. D. Varnado, A. G. Tennyson, D. M. Khramov, J. W. Kamplain, D. H. Sung, P. T. Cresswell, V. M. Lynch and C. W. Bielawski, Organometallics, 2009, 28, 6695–6706 CrossRef CAS.
- Y. Ryu, G. Ahumada and C. W. Bielawski, Chem. Commun., 2019, 55, 4451–4466 RSC.
- (a) C. Ruiz-Zambrana, A. Gutierrez-Blanco, S. Gonell, M. Poyatos and E. Peris, Angew. Chem., Int. Ed., 2021, 60, 20003–20011 CrossRef CAS PubMed; (b) C. Ruiz-Zambrana, R. K. Dubey, M. Poyatos, A. Mateo-Alonso and E. Peris, Chem. – Eur. J., 2022, 28, e202201384 CrossRef CAS PubMed.
- C. Ruiz-Zambrana, M. Poyatos and E. Peris, ACS Catal., 2022, 12, 4465–4472 CrossRef CAS.
- P. K. R. Panyam and T. Gandhi, Adv. Synth. Catal., 2017, 359, 1144–1151 CrossRef CAS.
- A. G. Tennyson, R. J. Ono, T. W. Hudnall, D. M. Khramov, J. A. V. Er, J. W. Kamplain, V. M. Lynch, J. L. Sessler and C. W. Bielawski, Chem. – Eur. J., 2010, 16, 304–315 CrossRef CAS PubMed.
- (a) G. Mancano, M. J. Page, M. Bhadbhade and B. A. Messerle, Inorg. Chem., 2014, 53, 10159–10170 CrossRef CAS PubMed; (b) B. Y. W. Man, M. Bhadbhade and B. A. Messerle, New J. Chem., 2011, 35, 1730–1739 RSC; (c) S. Elgafi, L. D. Field and B. A. Messerle, J. Organomet. Chem., 2000, 607, 97–104 CrossRef CAS; (d) Y. Huang, X. H. Zhang, X. Q. Dong and X. M. Zhang, Adv. Synth. Catal., 2020, 362, 782–788 CrossRef CAS; (e) M. J. Geier, C. M. Vogels, A. Decken and S. A. Westcott, Eur. J. Inorg. Chem., 2010, 4602–4610 CrossRef CAS; (f) A. M. Haydl and B. Breit, Chem. – Eur. J., 2017, 23, 541–545 CrossRef CAS PubMed; (g) A. Lumbroso, N. Abermil and B. Breit, Chem. Sci., 2012, 3, 789–793 RSC; (h) B. Y. W. Man, A. Knuhtsen, M. J. Page and B. A. Messerle, Polyhedron, 2013, 61, 248–252 CrossRef CAS.
- (a) A. Gutierrez-Blanco, E. Peris and M. Poyatos, Organometallics, 2018, 37, 4070–4076 CrossRef CAS; (b) E. Mas-Marza, E. Peris, I. Castro-Rodriguez and K. Meyer, Organometallics, 2005, 24, 3158–3162 CrossRef CAS; (c) M. Viciano, E. Mas-Marza, M. Sanau and E. Peris, Organometallics, 2006, 25, 3063–3069 CrossRef CAS; (d) E. Mas-Marza, M. Sanau and E. Peris, Inorg. Chem., 2005, 44, 9961–9967 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, NMR, UV-Vis and Mass spectra. Catalytic studies. CCDC 2170148. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc02497g |
| This journal is © The Royal Society of Chemistry 2022 |