
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diverse reactivity of an Al(I)-centred anion towards ketones†
Han-Ying
Liu
,
Michael S.
Hill
 * and
Mary F.
Mahon
* and
Mary F.
Mahon
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk
First published on 24th May 2022
Abstract
The reactivity of a seven-membered cyclic potassium diamidoalumanyl toward a variety of ketone small molecules has been assessed. Whilst acetophenone generates an aluminium pinacolate derivative, reductive C–C coupling is induced between the ketyl and ortho-carbon centres of two equivalents of benzophenone. In contrast, whereas oxidative addition of an enolisable proton is observed with 2,4-dimethyl-3-pentanone, 2,2,4,4-tetramethyl-3-pentanone undergoes an unprecedented hydroalumination process, where the reducing hydride may be traced to intramolecular oxidative addition of a (sp3)C–H bond.
Addition of a carbon-centred nucleophile to the carbonyl function of an aldehyde or ketone provides a textbook demonstration of C–C bond formation and the synthesis of secondary and tertiary alkoxide and, after hydrolysis, alcohol molecules.1,2 Although analogous reactivity with organosilyl anions can afford comparable C–Si bond formation,3 these latter processes are commonly complicated by (Brook-type) rearrangement to silyl ethers and related species driven by the thermodynamic preference for O–Si bond formation.4 A similar diversity of outcome has also started to emerge from the more recent and still limited reports of the reactivity of group 13-centred anions with carbonyl-containing molecules.5 Whereas, for example, Yamashita and co-workers’ nucleophilic lithium boryl, [(HCNDipp)2BLi] (Dipp = 2,6-di-iso-propylphenyl), was shown to attack the carbonyl group of benzaldehyde to afford the corresponding α-borylbenzyl alcohol,6 analogous treatment with benzophenone resulted in a ‘bora-Brook’ rearrangement via carbon-to-oxygen boryl migration.7 In a similar vein, we have previously observed that reaction of the β-diketiminate-supported terminal magnesium boryl [(BDI)Mg(Bpin)(DMAP)] (BDI = HC{(Me)CNDipp}2; DMAP = 4-dimethylaminopyridine) with benzophenone provided the magnesium C-borylalkoxide, [(BDI)Mg{OCPh2(Bpin)}(DMAP)], resulting from nucleophilic addition of the [Bpin]− anion at the carbonyl carbon.8 In contrast, similar treatment of its diboranate precursor, [(BDI)Mg{pinB-B(n-Bu)pin}] (1), which can act as a surrogate for the [Bpin]− unit in its own right, with benzophenone provided phenyl dearomatisation and compound 2, which subsequently induces nucleophilic attack at the carbonyl carbon centre of a further equivalent of benzophenone to afford 3 (Scheme 1a).9
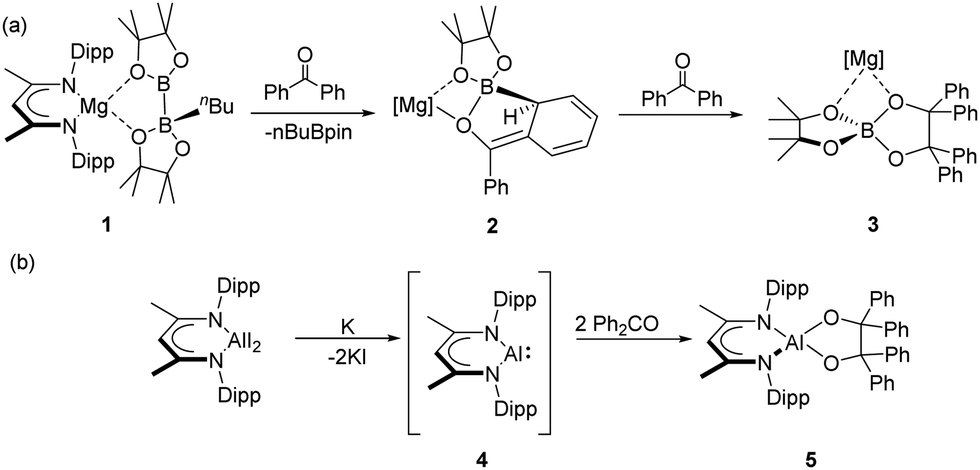 | ||
| Scheme 1 Selected examples of reductive coupling of benzophenone mediated by low-oxidation state group 13 centres. | ||
Although the structure of 2 argued for a mechanism derived from enolate attack at a further carbonyl equivalent, this latter behaviour is also reminiscent of the alkali or alkaline-earth metal-mediated coupling of benzophenone and 9-fluorenone ketyl radicals.10,11 A more explicit demonstration of this latter form of benzophenone coupling had earlier been provided by its reaction with an in situ generated source of the neutral Al(I) species, [(BDI)Al(I)] (4),12 which yielded a pinacolatoaluminium(III) derivative (5) (Scheme 1b).13 More recent developments have seen the emergence of a further class of Al(I) species in which a formally anionic aluminium centre is supported by a variety of diamido-, dialkyl-, and alkylamido- backbones (6–10) (Fig. 1).14–19 Although a rich reactivity for such alumanyl species has begun to emerge,5b their behaviour toward ketonic molecules as archetypal carbon electrophiles has not yet been described.20 In this contribution we begin to address this lacuna through a study of the reactivity of the seven-membered heterocyclic potassium diamidoalumanyl (8) toward a variety of ketone small molecules.
 | ||
| Fig. 1 Selected examples of molecules comprising an anionic Al(I) centre.14–19 | ||
Intrigued by the related but contrasting modes of reactivity displayed by 1 and 4 toward benzophenone, [K{Al(SiNDipp)}]2 (8, where SiNDipp = {CH2SiMe2NDipp}2) was reacted with four molar equivalents of benzophenone, resulting in a bright yellow oil and colourless supernatant. Addition of a few drops of tetrahydrofuran (THF) provided a homogeneous yellow solution, from which crystals suitable for X-ray crystallographic analysis were obtained by slow evaporation at room temperature. Surprisingly, the product from the reaction was not a symmetrically-disposed tetraphenylpinacolate derivative analogous to that obtained from the reaction between 4 and benzophenone. Rather, the resultant molecule, K(THF)2[(SiNDipp)Al-κ2-O,O′-{OCPh2CH(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCHCH)CCPhO}] (11), is a molecular, spirocyclic aluminate comprising a pair of seven-membered heterocycles (Al1–N1–Si1–C15–C16–Si2–N2 and Al1–O1–C31–C32–C33–C34–O2) (Scheme 2 and Fig. 2a), with charge balance maintained through encapsulation of K1 by an enolate-like {C33–C34–O2} subunit, an η6-coordinated N-Dipp substituent and two molecules of THF.
CHCHCH)CCPhO}] (11), is a molecular, spirocyclic aluminate comprising a pair of seven-membered heterocycles (Al1–N1–Si1–C15–C16–Si2–N2 and Al1–O1–C31–C32–C33–C34–O2) (Scheme 2 and Fig. 2a), with charge balance maintained through encapsulation of K1 by an enolate-like {C33–C34–O2} subunit, an η6-coordinated N-Dipp substituent and two molecules of THF.
 | ||
| Scheme 2 Reaction of [K{Al(SiNDipp)}]2 (8) with benzophenone and acetophenone to provide 11 and 12, respectively. | ||
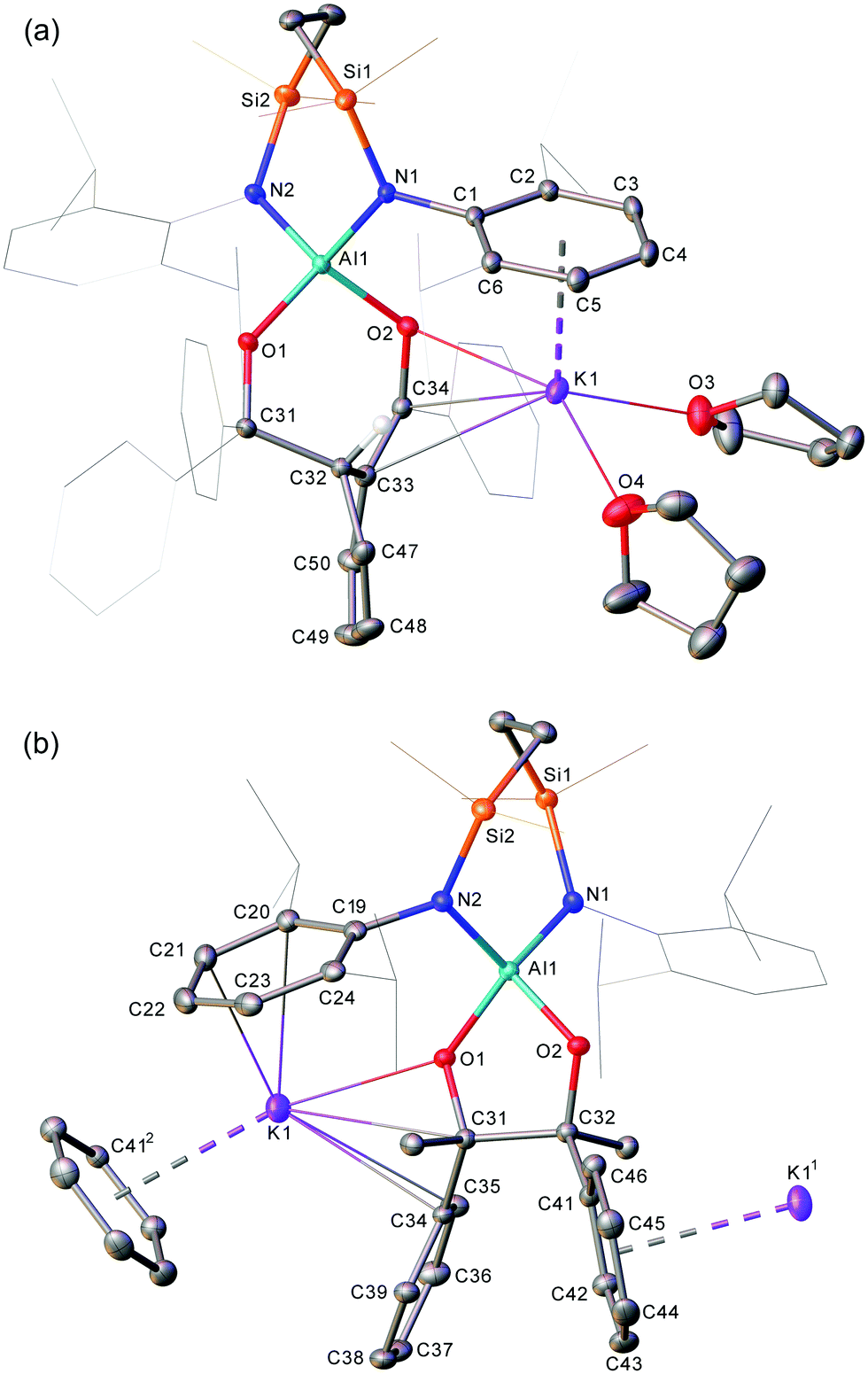 | ||
| Fig. 2 Displacement ellipsoid (30% probability) plots of (a) compound 11 and (b) a polymeric section of compound 12. For purposes of clarity, hydrogen atoms, apart from that attached to C32 in 11, are omitted, and selected aryl substituents are shown as wireframe. Selected bond distances (Å) and bond angles (°): (11) Al1–O1 1.8008(12), Al1–O2 1.7700(12), Al1–N1 1.8705(15), Al1–N2 1.8855(15), O1–C31 1.437(2), O2–C32 1.408(2) C31–C32 1.607(2), O1–Al1–O2 91.52(6), N1–Al1–N2 111.67(7). (12) Al1–O2 1.7962(16), Al1–O1 1.7477(15), Al1–N2 1.8729(19), Al1–N1 1.8934(18), O1–C31 1.400(2), C31–C32 1.596(3), C32–C33 1.529(3), C33–C34 1.362(3), O2–C34 1.353(3), O1–Al1–O2 105.11(7), N1–Al1–N2 109.56(8). | ||
Although, like 5, the structure of 11 represents a product of benzophenone reductive coupling, C–C bond formation has occurred between a single ketyl carbon and an ortho-phenyl carbon of a second benzophenone moiety, resulting in a dearomatised six-membered carbocycle. Consistent with an assignment of a now formal Al(III) oxidation state, both the Al–N [1.8729(19) and 1.8934(18) Å] and Al–O [1.7962(16) and 1.7477(15) Å] distances in 11 are comparable to those of 5, while, reminiscent of the structure of 2, the alternating long [C32–C33 1.529(3), C32–C47 1.516(3) and C48–C49 1.453(4) Å] and short [C47–C48 1.335(4), C49–C50 1.346(4) Å] C–C bonds clearly denote a dienic structure to the dearomatised unit. Solution-state characterisation of 11 conducted in d8-THF provided a 1H NMR spectrum comprising individual resonances characteristic of the dearomatized ring at δ 6.04, 5.60, 5.21, 5.10 and 3.69 ppm, which were assigned to the respective protons on the carbon atoms identified in the solid–state structure as C50, C47, C48, C49 and C32, respectively. These signals were observed alongside four discriminated SiMe peaks at δ 0.73, 0.46, −0.45, −0.55 ppm, each 3H by relative integration (Fig. S1, ESI†).
A reaction was then performed between 8 and four equivalents of acetophenone. This resulted in decolourisation of the initial bright yellow solution and the precipitation of a colourless powder. Although in situ spectroscopic characterisation of this product (12) was hampered by its poor solubility, a likely result of its polymeric structure, its identity was confirmed by crystallographic characterisation of single crystals obtained by slow cooling of reactions performed at 60 °C, which were found to provide identical unit cells, irrespective of whether they were performed in d6-benzene or protio-toluene (Scheme 2 and Fig. 2b). The Al–N [1.8705(15), 1.8855(15) Å] and Al–O [1.8008(12) and 1.7700(12) Å] bond distances in compound 12 are again commensurate with oxidation to Al(III). In this case, however, and in a fashion reminiscent of the tetraphenylpinacolate species (5),13 the reaction provides more conventional reductive ketyl C–C coupling through the carbon centres of the previous carbonyl carbon centres and the generation of a polymeric structure propagated through a sequence of intra- and intermolecular polyhapto phenyl-to-potassium and Dipp-to-potassium bridging interactions.
To further shed light on how the reductive reactivity of anionic Al(I) is affected by the identity of the ketone reagent, compound 8 was then reacted with 2,4-dimethyl-3-pentanone. Colourless single crystals were obtained from a reaction in d6-benzene heated at 60 °C for eight hours. The resultant X-ray analysis identified the reaction product as a further polymeric derivative, the potassium (hydrido)(carboxyl)diamidoaluminate (13) (Scheme 3 and Fig. 3). Although we have not further assessed the mechanistic implications of the formation of 13, its structure represents the formal oxidative addition of an enolisable iso-propyl methine proton of 2,4-dimethyl-3-pentanone at the aluminium centre of 8, an inference supported by the C31–C32 [1.338(3) Å] and C31–C35 [1.518(3) Å] distances, which are clearly indicative of the enolate formulation of the resultant oxygen-donor anion.13,14,21 Solution-state characterisation of 13 revealed a distinctive splitting of the SiMe2 environments at δ −0.05 and −0.09 ppm (each 6H by integration) in the 1H NMR spectrum along with a diagnostic 13C resonance at 94.7 ppm assigned as the newly formed tertiary sp2 (C32) carbon.
 | ||
| Scheme 3 Synthesis of compound 13. | ||
 | ||
| Fig. 3 Displacement ellipsoid (30% probability) plot of a polymeric section of compound 13. For purposes of clarity, most hydrogen atoms, disordered atoms and occluded benzene solvent are omitted, and selected methyl and iso-propyl substituents are shown as wireframe. Selected bond distances (Å) and bond angles (°): Al1–O1 1.7329(13), Al1–N2 1.8722(13), Al1–N1 1.8779(14), O1–C31 1.348(2), C31–C32 1.338(3), O1–Al1–N2 114.23(6), O1–Al1–N1 107.12(6), N2–Al1–N1 111.71(6). | ||
Prompted by the formation of 13, but to avoid the presence of a potentially reactive enolisable proton, 2,2,4,4-tetramethyl-3-pentanone was then selected as a further ketone substrate to be reacted with 8. Colourless single crystals of compound 14 formed direct from the reaction mixture after heating in toluene for 12 hours at 60 °C. X-ray diffraction analysis revealed 14 to be a tetrameric potassium organo-alkoxyaluminate comprising two independent aluminium-containing molecules within the unit cell, which are interconnected into a cyclotetrameric aggregate by a sequence of potassium–aryl interactions (Scheme 4 and Fig. 4). The 2,2,4,4-tetramethyl-3-pentanoxy substituents [Al1–O1 1.7508(15); Al2–O2 1.7553(12) Å] have evidently arisen from formal hydroalumination of 2,2,4,4-tetramethyl-3-pentanone, such that the former sp2 carbonyl carbon centres now adopt tetrahedral geometries [O1–C31 1.422(3), O2–C70 1.419(2) Å]. The hydrogen atoms associated with this reductive process can be deduced to have originated from intramolecular activation of a C–H bond of the Dipp iso-propyl methyl substituents, such that the fourth coordination sites of both independent aluminate anions are provided by newly formed [Al1–C 30 2.0214(18); Al2–C69 2.0159(17) Å] aluminium-to-methylene bonds.
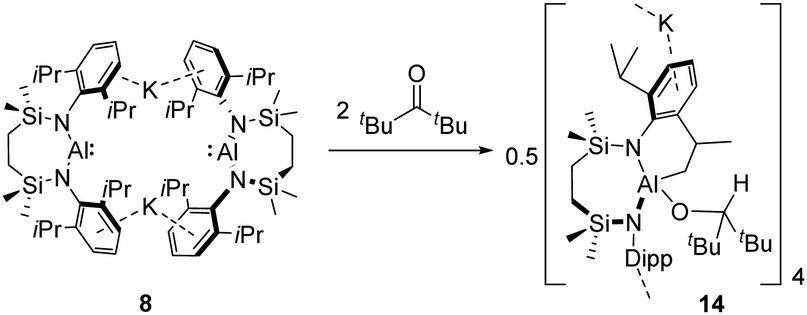 | ||
| Scheme 4 Synthesis of compound 14. | ||
 | ||
| Fig. 4 Displacement ellipsoid (30% probability) plot of the tetrameric aggregate of compound 14. For purposes of clarity, most hydrogen atoms are omitted, and selected methyl and iso-propyl substituents are shown as wireframe. Selected bond distances (Å) and bond angles (°): Al1–O1 1.7509(15), Al1–N2 1.9083(14), Al1–N1 1.9003(17), Al1–C30 2.0195(19), O1–C31 1.421(3), N1–Al1–N2 115.11(7), O1–Al1–C30 114.35(8), N1–Al1–C30 104.91(8), N2–Al1–C30 99.95(7), O1–C31–C36 110.45(17), O1–C31–C32 109.02(18), C32–C31–C36 118.93(18). | ||
Although further solution-state analysis of this process was precluded by the simultaneous formation and crystallisation of 14 under the applied reaction conditions, this reactivity plausibly ensues by initial coordination of a molecule of ketone to the aluminium centre of 8. Previous studies of compound 6 have highlighted the augmented reductive Al(I) reactivity toward the C–C bonds of even benzene solvent that may be induced by abstraction of its potassium cations with [2.2.2]cryptand.22 We, thus, surmise that similar disruption of its robust dimeric structure initiates the sterically-induced oxidative addition of a C(sp3)–H bond at the Al(I) centre of 8 to form the six-membered (Al1–N2–C19–C24–C28–C30 and Al2–N4–C58–C63–C67–C69) heterocycles. The transiently formed hydridic Al–H bonds subsequently attack the carbonyl carbon, to yield the observed alkoxide anions.23
The molecular structure of compound 14 discloses that each molecular unit possesses two stereogenic centres (Al1 and C31/Al2 and C70), the presence of which are also apparent by NMR spectroscopy. The 1H NMR spectrum, which was necessarily recorded in more coordinating d8-THF (Fig. S15, ESI†), demonstrates that there are two diastereomers, which, although inseparable are clearly distinguishable. Most characteristically, two singlets at δ 3.63 ppm and 2.75 ppm may be unambiguously attributed to the Al–O–CH environments of each diastereomer, an assignment verified by a 1H–13C HSQC experiment (Fig. S18, ESI†). The consequent asymmetry of the SiNDipp backbone was also clearly evident from the observation of four unique environments, each 3H by relative integration, for the silylmethyl substituents.
In conclusion, the potassium alumanyl 8 has been observed to offer a variety of substrate-dependent outcomes when reacted with ketone small molecules. Whilst the reaction of 8 with acetophenone generates the aluminium pinacolate derivative in a manner reminiscent of that previously observed for charge neutral Al(I) species, an unconventional reductive C–C coupling is induced between the ketyl and ortho-carbon centres of two equivalents of benzophenone. In contrast, whereas oxidative addition of an enolisable proton is observed with 2,4-dimethyl-3-pentanone, 2,2,4,4-tetramethyl-3-pentanone undergoes an unprecedented hydroalumination process, where the reducing hydride may be rationalised as resulting from intramolecular oxidative addition of a (sp3)C–H bond.
H.-Y. L. performed the synthesis and characterisation of the new compounds reported and prepared an initial draft of the manuscript. MSH conceptualised the study and finalised the manuscript for submission. MFM finalised the X-ray diffraction studies for publication.
We thank the EPSRC (UK) for their generous support of this research (EP/R020752/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Schlosser, Organometallics, in Synthesis: A Manual, Wiley, Chichester, 2nd Edn, 2001 Search PubMed.
- Reviews for secondary and tertiary alcohol synthesis: (a) C. García and V. S. Martín, Curr. Org. Chem., 2006, 10, 1849–1889 CrossRef; (b) M. Hatano, T. Miyamoto and K. Ishihara, Curr. Org. Chem., 2007, 11, 127 CrossRef CAS; (c) O. Riant and J. Hannedouche, J. Org. Biomol. Chem., 2007, 5, 873–888 RSC; (d) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2007, 5969–5994 CrossRef CAS; (e) M. Hatano and K. Ishihara, Synthesis, 2008, 1647–1675 CAS.
- A. G. Brook, J. Am. Chem. Soc., 1958, 80, 1886–1889 CrossRef CAS.
- See, for example: P. Cuadrado, A. M. González, B. González and F. J. Pulido, Synth. Commun., 1989, 19, 275–283 CrossRef CAS.
- For relevant reviews, see: (a) M. Yamashita, Bull. Chem. Soc. Jpn., 2011, 84, 983–999 CrossRef CAS; (b) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS PubMed.
- (a) Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS PubMed; (b) Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 CrossRef CAS PubMed.
- H. Kisu, H. Sakaino, F. Ito, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2016, 138, 3548–3552 CrossRef CAS PubMed.
- A.-F. Pécharman, A. L. Colebatch, M. S. Hill, C. L. McMullin, M. F. Mahon and C. Weetman, Nat. Commun., 2017, 8, 15022 CrossRef PubMed.
- A.-F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Organometallics, 2018, 37, 4457–4464 CrossRef.
- H. Gilman and R. E. Brown, J. Am. Chem. Soc., 1930, 52, 1128–1131 CrossRef CAS.
- Z. Hou, X. Jia, A. Fujita, H. Tezuka, H. Yamazaki and Y. Wakatsuki, Chem. – Eur. J., 2000, 6, 2994–3005 CrossRef CAS PubMed.
- C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS.
- (a) C. Cui, S. Köpke, R. Herbst-Irmer, H. W. Roesky, M. Noltemeyer, H.-G. Schmidt and B. Wrackmeyer, J. Am. Chem. Soc., 2001, 123, 9091–9098 CrossRef CAS PubMed ; for a general review of oxidative addition at main group element centres, see: ; (b) T. Chu and G. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed.
- R. J. Schwamm, M. P. Coles, M. S. Hill, M. F. Mahon, C. L. McMullin, N. A. Rajabi and A. S.-S. Wilson, Angew. Chem., Int. Ed., 2020, 59, 3928–3932 CrossRef CAS PubMed.
- S. Kurumada, S. Takamori and M. Yamashita, Nat. Chem., 2020, 12, 36–39 CrossRef CAS PubMed.
- K. Koshino and R. Kinjo, J. Am. Chem. Soc., 2020, 142, 9057–9062 CrossRef CAS PubMed.
- S. Grams, J. Eyselein, J. Langer, C. Farber and S. Harder, Angew. Chem., Int. Ed., 2020, 59, 15982–15986 CrossRef CAS PubMed.
- Interesting reactivity of alumanyl anions with the CO bonds of CO2, isocyanates and CO has, however, begun to emerge, see:
(a) J. Hicks, A. Heilmann, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 17265–17268 CrossRef CAS PubMed;
(b) M. J. Evans, M. G. Gardiner, M. D. Anker and M. P. Coles, Chem. Commun., 2022, 58, 5833–5836 RSC.
- For related examples of O–H and N–H oxidative addition to an alumanyl anion, see; M. J. Evans, M. D. Anker and M. P. Coles, Inorg. Chem., 2021, 60, 4772–4778 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS PubMed.
- For a related example of iso-propyl C–H bond activation at the AlO bond of an alumoxane derived from a potassium diamidoalumanyl, see; M. D. Anker and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 18261–18265 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic experimental details, NMR spectra, details of the X-ray analysis of compounds 11–14. CCDC 2167478–2167481. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc02333d |
| This journal is © The Royal Society of Chemistry 2022 |