
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of cyclic α-1,4-oligo-N-acetylglucosamine ‘cyclokasaodorin’ via a one-pot electrochemical polyglycosylation–isomerization–cyclization process†
Hirofumi
Endo
a,
Masaharu
Ochi
a,
Md Azadur
Rahman
a,
Tomoaki
Hamada
b,
Takahiro
Kawano
b and
Toshiki
Nokami
 *ac
*ac
aDepartment of Chemistry and Biotechnology, Tottori University, 4-101 Koyamacho Minami, Tottori City, 680-8552 Tottori, Japan. E-mail: tnokami@tottori-u.ac.jp
bKoganei Corporation, 3-11-28 Midorimachi, Koganei City, 184-8533 Tokyo, Japan
cCentre for Research on Green Sustainable Chemistry, Faculty of Engineering, Tottori University, 4-101 Koyamacho Minami, Tottori City, 680-8552 Tottori, Japan
First published on 24th June 2022
Abstract
Electrochemical synthesis of unnatural cyclic oligosaccharides composed of N-acetylglucosamine with α-1,4-glycosidic linkages has been accomplished. A thioglycoside monomer equipped with the 2,3-oxazolidinone protecting group was used to prepare linear oligosaccharides by electrochemical polyglycosylation. In the same pot, isomerization of the linear oligosaccharides and intramolecular electrochemical glycosylation for cyclization were also conducted sequentially to obtain the precursor of the cyclic α-1,4-oligo-N-acetylglucosamine ‘cyclokasaodorin’.
Electrochemical transformations of organic molecules have attracted significant attention over the last decade.1 Both anodic oxidation and cathodic reduction can generate highly reactive chemical species, including strong acid and base under mild reaction conditions, and their amount can be precisely controlled under the constant current condition. These beneficial properties of electrochemical methods enable unique molecular transformations that are difficult to achieve using conventional chemical methods.2
We have been interested in the synthesis of oligosaccharides under electrochemical conditions.3 Cyclic oligosaccharides are one example of electrochemically synthesized oligosaccharides; however, it required the preparation of linear oligosaccharides to convert them to the corresponding cyclic oligosaccharides.4 Chemical synthesis of a variety of cyclic oligosaccharides has already been reported; however, multiple-step synthesis had to be performed using conventional methods of glycosylation.5 Thus, an ideal approach to synthesizing cyclic oligosaccharides is glycosylation polymerization (i.e., polyglycosylation). Indeed, the synthesis of cyclic oligosaccharides under the polyglycosylation conditions has already been reported; however, it is limited to β-1,6-glycosidic linkages.6
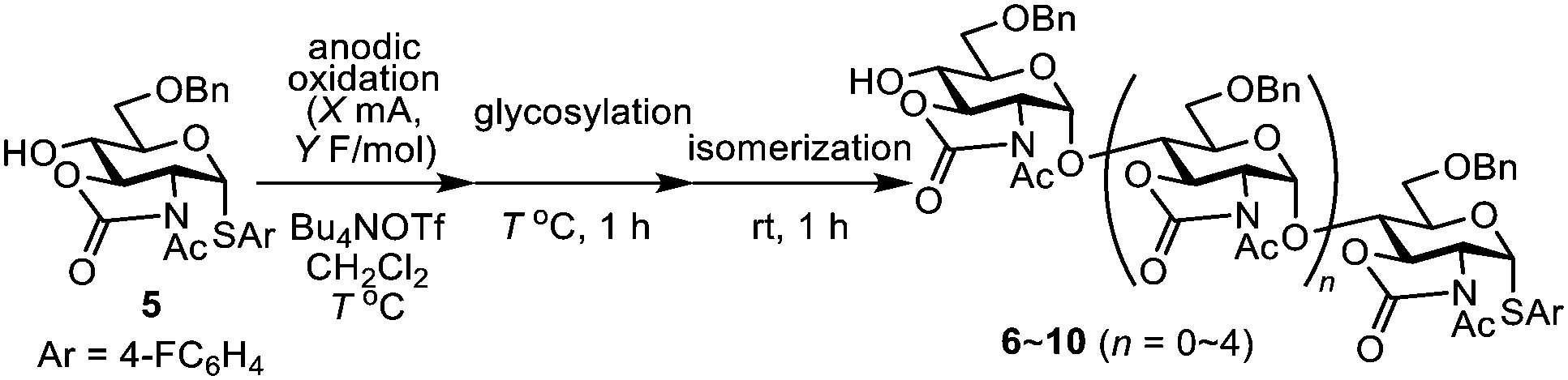
Based on this background, we planned the retrosynthesis of the unnatural cyclic α-1,4-hexa-N-acetylglucosamine 1 (n = 1) which has never been reported previously (Scheme 1). Cyclic oligosaccharides 2 (n = 1), 3 (n = 2), and 4 (n = 3) with protecting groups will be precursors of the desired cyclic hexasaccharide 1 (n = 1) and larger oligosaccharides. These precursors might be synthesized by polyglycosylation of thioglycoside 5, which is a monosaccharide derived from glucosamine hydrochloride (Scheme 1). Thioglycoside 5 was chosen as a monomer because thioglycosides with the 2,3-oxazolidinone protecting group have been used extensively for the α-selective synthesis of glucosamines via acid-catalysed isomerization.7 We assumed that the polyglycosylation of thioglycoside 5 might be achieved using a one-pot three-step process including two steps of electrochemical oxidation and an isomerization step (Scheme 2). In situ generation of linear oligosaccharides 6–10 with α-1,4-glycosidic bonds are crucial for the success of this process. Here, we report the first total synthesis of cyclic α-1,4-hexa-N-acetylglucosamine 1.
 | ||
| Scheme 1 Retrosynthesis of cyclic α-1,4-oligo-N-acetylglucosamine. | ||
 | ||
| Scheme 2 Electrochemical polyglycosylation–isomerization–cyclization for the synthesis of cyclic oligosaccharides. | ||
We initiated our study for the development of the novel one-pot electrochemical process from the optimization of electrochemical polyglycosylation to prepare linear oligosaccharides 6–10 with α-1,4-glycosidic bonds (Table 1). Our previous study has already had shown that the isomerization from the β-isomer to α-isomer of glycosides occurred within 1 h above 0 °C.7f This isomerization process was promoted by in situ-generated triflic acid (TfOH) using tetrabutylammonium triflate (Bu4NOTf) as an electrolyte. Thus, we decided to raise the reaction temperature from T °C to room temperature (rt) after electrochemical polyglycosylation to accelerate the isomerization process, although it took time to control the temperature of the reaction.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Conditions | Yields of oligosaccharides | ||||||
| X mA | Y F mol−1 | T °C | 6 n = 0 (%) | 7 n = 1 (%) | 8 n = 2 (%) | 9 n = 3 (%) | 10 n = 4 (%) | |
| a Average of 2 runs. | ||||||||
| 1 | 8 | 0.6 | –20 | 14 | 21 | 16 | 11 | 7.4 |
| 2a | 8 | 0.6 | –30 | 21 | 24 | 16 | 7.5 | 7.2 |
| 3a | 8 | 0.6 | –40 | 21 | 18 | 13 | 8.7 | 7.7 |
| 4 | 8 | 0.6 | –50 | 18 | 20 | 11 | 11 | 6.6 |
| 5 | 8 | 0.6 | –60 | 17 | 20 | 13 | 8.9 | 4.6 |
| 6 | 8 | 0.7 | –40 | 13 | 18 | 15 | 8.3 | 7.6 |
| 7 | 4 | 0.6 | –40 | 22 | 19 | 13 | 8.9 | 6.3 |
| 8 | 12 | 0.6 | –40 | 25 | 22 | 14 | 8.1 | 5.5 |
The reaction temperature for electrochemical polyglycosylation was examined from –20 °C to –60 °C (entries 1–5). Yields of linear oligosaccharides 6–10 (n = 0 to 4) were determined after isolation by preparative gel permeation chromatography (GPC). Yields of oligosaccharides were better at higher reaction temperatures, such as –20 °C and –30 °C (entries 1 and 2); however, oligosaccharides of hydroxy sugars, which have a hydroxy group instead of the thioaryl (SAr) group at the anomeric carbon, were observed as by-products. Desired linear hexasaccharide 10 (n = 4), which was a potential precursor of cyclic oligosaccharide 2, was obtained up to 7.7% at temperatures below –40 °C (entries 3–5). Increasing electricity from 0.6 F mol−1 to 0.7 F mol−1 did not improve the yields of oligosaccharides (entry 6) and further investigation of the current value was not effective to obtain oligosaccharides in better yields (entries 7 and 8). Although the corresponding peaks of linear heptasaccharide and octasaccharide were observed using matrix assisted laser desorption/ionization time of flight mass spectroscopy (MALDI-TOF MS), they were very difficult to isolate in high purity. Unreacted thioglycoside 5 was recovered, and formation of 1,4-anhydrosugar, which must be generated by intramolecular glycosylation, was not observed in this reaction.
Conversion of linear hexasaccharide 10 (n = 4) to cyclic hexasaccharide 2 (n = 1) was also investigated under the anodic oxidation conditions at –40 °C (Scheme 3). Although it required an excessive amount of electricity (3.0 F mol−1), linear hexasaccharide 10 (n = 4) was completely consumed (>99%). Furthermore, the corresponding cyclic hexasaccharide 2 was obtained in 44% yield. This was an important result for the development of the one-pot electrochemical process because the reaction conditions of the electrochemical cyclization were almost the same as those of electrochemical polyglycosylation (Table 1).
 | ||
| Scheme 3 Electrochemical cyclization of linear hexasaccharide. | ||
The isomerization step after electrochemical polyglycosylation is crucial to obtaining the precursor of a cyclic oligosaccharide with α-glycosidic linkages. The 1H NMR spectrum of linear hexasaccharide 10′ obtained without the isomerization step is shown in Fig. 1b together with thioglycoside 5, linear hexasaccharide 10, and cyclic hexasaccharide 2. The peaks between 4.8 and 5.5 ppm ( ) indicate the presence of β-glycosidic linkages in linear hexasaccharide 10′. The complexity of the peaks may stem from the difference in the number and position of β-glycosidic linkages of linear hexasaccharide 10′. After the isomerization of 10′ the corresponding peaks of the β-glycosidic linkages disappeared as shown in the spectrum of linear hexasaccharide 10, and two sets of peaks at 6.07 ppm (doublet, J = 4.4 Hz, 1 H,
) indicate the presence of β-glycosidic linkages in linear hexasaccharide 10′. The complexity of the peaks may stem from the difference in the number and position of β-glycosidic linkages of linear hexasaccharide 10′. After the isomerization of 10′ the corresponding peaks of the β-glycosidic linkages disappeared as shown in the spectrum of linear hexasaccharide 10, and two sets of peaks at 6.07 ppm (doublet, J = 4.4 Hz, 1 H,  ) and 6.03–6.00 ppm (multiplet, 5 H,
) and 6.03–6.00 ppm (multiplet, 5 H,  ) were observed around 6.0 ppm (Fig. 1c). The peak at 6.07 ppm was assigned to the anomeric proton of the reducing end of the oligosaccharide 10 because it was in good agreement with the anomeric proton of thioglycoside 5 at 6.05 ppm (doublet, J = 4.5 Hz, 1 H,
) were observed around 6.0 ppm (Fig. 1c). The peak at 6.07 ppm was assigned to the anomeric proton of the reducing end of the oligosaccharide 10 because it was in good agreement with the anomeric proton of thioglycoside 5 at 6.05 ppm (doublet, J = 4.5 Hz, 1 H,  ). After cyclization of 10, no peaks were observed at 6.0 ppm, and the doublet peak at 5.80 ppm (J = 2.3 Hz, 6 H,
). After cyclization of 10, no peaks were observed at 6.0 ppm, and the doublet peak at 5.80 ppm (J = 2.3 Hz, 6 H,  ) was assigned to the α-glycosidic linkage of cyclic hexasaccharide 2 (Fig. 1d).
) was assigned to the α-glycosidic linkage of cyclic hexasaccharide 2 (Fig. 1d).
 | ||
| Fig. 1 Comparison of 1H NMR spectra. (a) thioglycoside 5, (b) linear hexasaccharide without isomerization 10′, (c) linear hexasaccharide 10, (d) cyclic hexasaccharide 2. | ||
Based on these results, we performed a one-pot electrochemical polyglycosylation–isomerization–cyclization (ePIC) process using our automated electrochemical synthesizer (Scheme 4).8 Electrochemical polyglycosylation starts at –40 °C, and the reaction mixture was stirred for another 1 h at the same temperature (–40 °C). In the second step, isomerization was promoted at rt for 1 h. The final electrochemical cyclization was again performed at –40 °C under the same anodic oxidation conditions. Under the optimized conditions of each step of the process, the targeted cyclic oligosaccharides 2 (n = 1) and 3 (n = 2) were obtained in yields of 6.2% and 5.5%, respectively, together with a trace amount of 4 (n = 3). Although the size difference of cyclic oligosaccharides 2-4 was difficult to distinguish by NMR spectra, molecular ion peaks in MALDI-TOF MS spectra clearly showed 319 Da difference which was the molecular weight of the repeating unit. Larger cyclic oligosaccharide 4 (n = 3) was more polar than smaller oligosaccharides 2 (n = 1) and 3 (n = 2) as determined by TLC; however, we used a preparative GPC for purification.
 | ||
| Scheme 4 Preparation of cyclic oligosaccharides via the ePIC process. | ||
To confirm the advantage of the ePIC process, we demonstrated the single-step anodic oxidation under the optimized reaction temperature with 1.1 F mol−1 of electricity for comparison (Scheme 5). Although we could isolate cyclic hexasaccharide 2 (n = 1) and heptasaccharide 3 (n = 2), the yields were determined using 1H NMR with 1,1,2,2-tetrachloroethane as an internal standard because of the low yield and purity of the products. The formation of cyclic octasaccharide 4 (n = 3) was not detected (ND), even when using MALDI-TOF MS. We also performed the anodic oxidation with an increasing amount of electricity (1.8 F mol−1); however, the corresponding cyclic oligosaccharides 2–4 were not obtained. These results suggested that the ePIC process has an advantage over the conventional single-step electrochemical polyglycosylation in obtaining the target cyclic oligosaccharides.
 | ||
| Scheme 5 Electrochemical polyglycosylation with increasing amount of electricity as a control experiment. | ||
Global deprotection of cyclic hexasaccharide 2 was started with 2,3-oxazolidinone ring-opening using ethanethiol (EtSH) in the presence of K2CO3 (Scheme 6).9 The subsequent oxidative removal of the thioester of 11 was achieved by the reaction of an acetone solution of dimethyldioxirane (DMDO) selectively, and the final deprotection of the benzyl group at the 6-position (6–OH) of intermediate 12 was achieved under the conventional hydrogenation reaction conditions using Pd(OH)2/C as a catalyst. The global deprotection of precursor 2 afforded the desired cyclic oligosaccharide 1 in 52% total yield (three steps). Further optimization of reaction conditions is in progress to achieve global deprotection of precursors 3 and 4.
 | ||
| Scheme 6 Global deprotection of the protected cyclic oligosaccharide. | ||
In conclusion, we have achieved the first total synthesis of cyclic-α-1,4-hexa-N-acetylglucosamines, which are the 2-deoxy-2-acetoamide analogues of α-cyclodextrin and a cyclic variant of chito-oligosaccharides. We call these cyclic oligoglucosamines ‘cyclokasaodorin’ which was inspired by the unnatural cyclic oligosaccharide ‘cycloawaodorin’10 reported from the Nishizawa group in Tokushima and the traditional umbrella dance ‘Kasaodori’ in Tottori. Further optimization and modification of the synthetic process for large-scale synthesis of cyclokasaodorin to investigate their physical properties are in progress in our laboratory.
We acknowledge financial supports from the JSPS (No. JP19K05714). This paper is dedicated to the late Professor Hidetoshi Yamada of Kwansei Gakuin University.
H. E. and T. N. organized the research; H. E., M. O., and A. R. synthesized and characterized compounds; T. H. and T. K. developed the automated electrochemical synthesizer; H. E. and T. N. principally wrote the manuscript according to the suggestion and discussions with all authors.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef CAS PubMed; (b) J. Yoshida, Y. Ashikari, K. Matsumoto and T. Nokami, J. Synth. Org. Chem., Jpn., 2013, 71, 1136 CrossRef CAS; (c) J. Yoshida, A. Shimizu, Y. Ashikari, T. Morofuji, R. Hayashi, T. Nokami and A. Nagaki, Bull. Chem. Soc. Jpn., 2015, 88, 763 CrossRef CAS; (d) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (e) J. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702 CrossRef CAS PubMed; (f) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415 CrossRef CAS PubMed.
- (a) W. Zhang and S. Lin, J. Am. Chem. Soc., 2020, 142, 20661 CrossRef CAS PubMed; (b) Y. Kawamata, K. Hayashi, E. Carlson, S. Shaji, D. Waldmann, B. J. Simmons, J. T. Edwards, C. W. Zapf, M. Saito and P. S. Baran, J. Am. Chem. Soc., 2021, 143, 16580 CrossRef CAS PubMed; (c) T. Shen and T. H. Lambert, Science, 2021, 371, 620 CrossRef CAS PubMed; (d) D. E. Holst, D. J. Wang, M. J. Kim, I. A. Guzei and Z. K. Wickens, Nature, 2021, 596, 74 CrossRef CAS PubMed; (e) Y. Liu, X. Tao, X. Yuan, J. Qiu, L. Kong, S. Ni, K. Guo, Y. Wang and Y. Pan, Nat. Commun., 2021, 12, 6745 CrossRef CAS PubMed; (f) C. H. Wang, W. Ma, X. Zheng, M. Xu, X. Qi and Q. Lu, J. Am. Chem. Soc., 2022, 144, 1389 CrossRef PubMed.
- (a) T. Nokami, K. Saito and J. Yoshida, Carbohydr. Res., 2012, 363, 1 CrossRef CAS PubMed; (b) S. Manmode, K. Matsumoto, T. Itoh and T. Nokami, Asian J. Org. Chem., 2018, 7, 1719 CrossRef CAS; (c) A. Shibuya and T. Nokami, Chem. Rec., 2021, 21, 2389 CrossRef CAS PubMed; (d) K. Yano, N. Sasaki, T. Itoh and T. Nokami, J. Synth. Org. Chem., Jpn., 2021, 79, 839 CrossRef CAS.
- S. Manmode, S. Tanabe, T. Yamamoto, N. Sasaki, T. Nokami and T. Itoh, ChemistryOpen, 2019, 8, 869 CrossRef CAS PubMed.
- (a) Y. Takahashi and T. Ogawa, Carbohydr. Res., 1987, 164, 277 CrossRef CAS; (b) N. K. Kochetkov, S. A. Nepogodiev and L. V. Backinowsky, Tetrahedron, 1990, 46, 139 CrossRef CAS; (c) S. Houdier and P. J. A. Vottero, Carbohydr. Res., 1993, 248, 377 CrossRef CAS PubMed; (d) G. Gattuso, S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1919 CrossRef CAS PubMed; (e) M. Wakao, K. Fukase and S. Kusumoto, J. Org. Chem., 2002, 67, 8182 CrossRef CAS PubMed; (f) D. Ikuta, Y. Hirata, S. Wakamori, H. Shimada, Y. Tomabechi, Y. Kawasaki, K. Ikeuchi, T. Hagimori, S. Matsumoto and H. Yamada, Science, 2019, 364, 674 CrossRef CAS PubMed; (g) K. Maiti, G. C. Samanta and N. Jayaraman, Arkivoc, 2021, iv, 113 Search PubMed.
- H. Someya, T. Seki, G. Ishigami, T. Itoh, Y. Saga, Y. Yamada and S. Aoki, Carbohydr. Res., 2020, 487, 107888 CrossRef CAS PubMed.
- (a) P. Wei and R. J. Kerns, J. Org. Chem., 2005, 70, 4195 CrossRef CAS PubMed; (b) M. Boysen, E. Gemma, M. Lahmann and S. Oscarson, Chem. Commun., 2005, 3044 RSC; (c) S. Manabe, K. Ishii and Y. Ito, J. Am. Chem. Soc., 2006, 128, 10666 CrossRef CAS PubMed; (d) S. Manabe, K. Ishii and Y. Ito, J. Org. Chem., 2007, 72, 6107 CrossRef CAS PubMed; (e) H. Satoh, S. Manabe, Y. Ito, H. P. Lüthi, T. Laino and J. Hutter, J. Am. Chem. Soc., 2011, 133, 5610 CrossRef CAS PubMed; (f) T. Nokami, A. Shibuya, Y. Saigusa, S. Manabe, Y. Ito and J. Yoshida, Beilstein J. Org. Chem., 2012, 8, 456 CrossRef CAS PubMed; (g) S. Manabe, H. Satoh, J. Hutter, H. P. Luthi, T. Laino and Y. Ito, Chem. – Eur. J., 2014, 20, 124 CrossRef CAS PubMed; (h) S. Manabe and Y. Ito, Beilstein J. Org. Chem., 2016, 12, 328 CrossRef CAS PubMed; (i) S. Manabe and Y. Ito, Pure Appl. Chem., 2017, 89, 899 CAS.
- T. Nokami, R. Hayashi, Y. Saigusa, A. Shimizu, C.-Y. Liu, K.-K. Mong and J. Yoshida, Org. Lett., 2013, 15, 4520 CrossRef CAS PubMed.
- R. Koinuma, K. Tohda, T. Aoyagi and H. Tanaka, Chem. Commun., 2020, 56, 12981 RSC.
- (a) M. Nishizawa, H. Imagawa, Y. Kan and H. Yamada, Tetrahedron Lett., 1991, 32, 5551 CrossRef CAS; (b) M. Nishizawa, H. Imagawa, K. Kubo, Y. Kan and H. Yamada, Synlett, 1992, 447 CrossRef CAS; (c) M. Nishizawa, J. Synth. Org. Chem., Jpn., 1993, 51, 631 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral data of new compounds. See DOI: https://doi.org/10.1039/d2cc02287g |
| This journal is © The Royal Society of Chemistry 2022 |