
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic activity for proton reduction by a covalent non-metal graphene–fullerene hybrid†
Demetrios D.
Chronopoulos‡
 a,
Christina
Stangel‡
b,
Magdalena
Scheibe
a,
Klára
Čépe
a,
Nikos
Tagmatarchis
*b and
Michal
Otyepka
*ac
a,
Christina
Stangel‡
b,
Magdalena
Scheibe
a,
Klára
Čépe
a,
Nikos
Tagmatarchis
*b and
Michal
Otyepka
*ac
aRegional Centre of Advanced Technologies and Materials, Czech Advanced Technology and Research Institute (CATRIN), Palacký University Olomouc, Šlechtitelů 27, Olomouc 779 00, Czech Republic. E-mail: michal.otyepka@upol.cz
bTheoretical and Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vassileos Constantinou Avenue, Athens 11635, Greece. E-mail: tagmatar@eie.gr
cIT4Innovations, VSB – Technical University of Ostrava, 17. listopadu 2172/15, 708 00 Ostrava-Poruba, Czech Republic
First published on 6th July 2022
Abstract
A non-metal covalent hybrid of fullerene and graphene was synthesized in one step via fluorographene chemistry. Its electrocatalytic performance for the hydrogen evolution reaction and durability was ascribed to intrahybrid charge-transfer phenomena, exploiting the electron-accepting properties of C60 and the high conductivity and large surface area of graphene.
Electrocatalytic water splitting into oxygen and hydrogen is one of the most promising processes for the sustainable production of hydrogen as a carbon-neutral fuel.1 The most effective electrocatalysts, which are commercially used in water electrolysis devices and fuel cells, are based on Pt and RuO2.2 The prohibitive scarcity and cost of the noble metals limit the future development of a hydrogen-based economy. Owing to their low price and earth abundance, non-precious metal-based electrocatalysts, mainly composed of transition metals, are employed as alternatives.3 However, all metal-based catalysts face challenges such as low selectivity, poor durability, agglomeration and environmental issues.4 Therefore, it is necessary to develop efficient, low cost and highly durable catalysts of the hydrogen evolution reaction (HER) to support the growth of the hydrogen economy.5–7 Besides efforts toward minimizing the HER overpotential, and increasing the reaction kinetics, catalyst electrical conductivity and stability,8,9 a new class of catalysts composed of lightweight and abundant elements must be developed. Towards this end, the development of carbon-based metal-free electrocatalysts has been launched, receiving remarkable attention.4
Remarkable electron acceptor properties of fullerenes have rendered them as great candidates for the synthesis of efficient multifunctional metal-free electrocatalysts.10 Heterostructures based on fullerene (mainly C60) and low dimensional nanomaterials are gaining increasing interest due to their unique physicochemical properties, such as large surface areas and great electronic and mechanical properties.11 These heterostructures appear as efficient noble-metal-free and low-cost electrocatalysts to compete with precious state-of-the-art noble-metal catalysts.12 Noticeably, a non-metal carbon-based electrocatalyst was prepared through the adsorption of C60 onto single-walled carbon nanotubes (SWCNTs), which exhibited excellent performance toward the HER, oxygen evolution reaction (OER) and oxygen reduction reaction (ORR). Evidently, intermolecular charge transfer between C60 and SWCNTs largely induced high electrocatalytic activities.13 Additionally, a bimetallic phosphide Ni–Co–P coupled with C60 presented excellent HER activity, and it was concluded that C60 improved the electrocatalytic activity for the HER by enhancing the surface roughness and electrical conductivity.14 In another study, a covalently linked graphene–C60 hybrid, which was prepared via ball-milling, was employed as an electrocatalyst toward the ORR,15 exhibiting higher catalytic activity than that of the individual species C60 and graphite. This finding was ascribed to the generation of a positively charged graphene basal plane, as a result of charge transfer to C60. Also, covalently linked CNT–C60 hybrids very efficiently catalyze the two-electron reduction of oxygen to H2O2 due to high electron mobility through the fullerene units, large surface area and covalent linkage between C60 and CNTs.16 Nevertheless, the use of C60 as a potential electrocatalyst is rare; hence, it is of greatest interest to further investigate its covalent conjugation onto graphene and scrutinize the electrocatalytic performance of the newly formed heterostructure for proton reduction toward hydrogen evolution.
In recent years, fluorographene (FG)17,18 has been employed as an effective precursor for the preparation of covalently modified graphene derivatives with high conductivity and functionalization degree,19,20 synthesizing metal-free electrocatalysts for the ORR as well.21–23 However, graphene derivatives produced by FG have not been studied in other electrocatalytic reactions. Moreover, from the synthetic point of view, the utilization of FG as a template for the hybridization with C60 has not been reported yet. Strikingly, the efficient intermolecular charge-transfer processes between graphene derivatives and fullerene nanocages are expected to result in a large number of active sites, which may favor electrocatalytic processes.
Herein, we exploited the high conductivity and large surface area of graphene derivatives produced by FGs with excellent electron acceptor properties of C60 to prepare a novel carbon-based, metal-free electrocatalyst for the HER. In this frame, a covalently linked graphene–fullerene hybrid (denoted as G–C60 henceforth) was synthesized via the chemistry of FGs and characterized by complementary spectroscopic, microscopy, and thermal analysis techniques. Additionally, the electrocatalytic activity of G–C60 for the HER was studied for the first time by performing linear sweep voltammetry (LSV) measurements and electrochemical impedance spectroscopy (EIS), showing significantly improved electrocatalytic activity in comparison with its precursors. The synergistic interactions between the formed graphene by FG and C60 promoted the formation of abundant active sites for electrocatalytic proton reduction.
Compounds bearing amino groups are widely employed as nucleophiles for the efficient covalent modification of FG. Thus, a fulleropyrrolidine derivative carrying a primary amino moiety as a nucleophile (C60-pyr-NH2)24 reacted with the electrophilic FG (Fig. 1).25 The reaction was conducted at 130 °C in dry 1,2-dichlorobenzene (o-DCB) under a nitrogen atmosphere. Owing to the cascade substitution and defluorination, G–C60 was successfully synthesized. The inertness of C60 to cause reductive defluorination in FG was also studied through a control experiment (see Section S1.3 and Fig. S1 in the ESI†).
 | ||
| Fig. 1 Preparation of the G–C60 hybrid via the chemistry of fluorographene. | ||
Successful covalent linking of fullerene derivative C60-pyr-NH2 onto the graphene lattice was proved by Fourier-transform infrared (FT-IR) spectroscopy (Fig. 2(A)). The FT-IR spectrum of the G–C60 hybrid displayed the aliphatic C–H stretching vibrations of the ethylene glycol chain at 2980–2850 cm−1 and characteristic vibrational peaks attributed to C60 at 1450, 1130 and 525 cm−1.26 Moreover, the significant reduction of the ascribed peak to the C–F bonds at 1200 cm−1 and the appearance of two bands at around 1585 and 1460 cm−1 verified the formation of the conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds due to the reductive defluorination of FG,27 which occurs simultaneously with the covalent grafting of fullerene derivative C60-pyr-NH2.
C double bonds due to the reductive defluorination of FG,27 which occurs simultaneously with the covalent grafting of fullerene derivative C60-pyr-NH2.
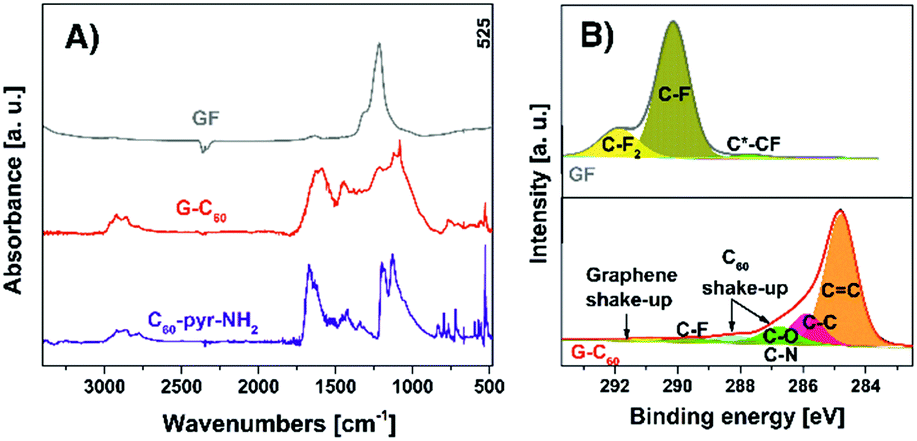 | ||
| Fig. 2 (A) FT-IR spectra of pristine GF, C60-pyr-NH2 and G–C60, and (B) HR-XPS C 1s spectra of pristine GF (upper panel) and G–C60 (lower panel). | ||
X-ray photoelectron spectroscopy (XPS) measurements also confirmed the successful synthesis of G–C60 providing information about the chemical composition of the material. According to atomic content analyses obtained from XPS, G–C60 showed an almost quantitative elimination of fluorine atoms (1.5 at% of F) in G–C60 with respect to 55.7 at% in graphite fluoride (GF) (Fig. S2 and Table S1, ESI†). In this context, the high-resolution C 1s XPS spectrum of G–C60 corroborated the nucleophilic substitution and reductive defluorination of FG, since the intensity of the C–Fx components at 288–293 eV practically disappeared (Fig. 2(B)). On the other hand, new bands appeared at 284.8 and 285.8 eV, ascribed to sp2 and sp3 carbons, respectively, indicating the formation of a graphenic network and the covalent linking of C60-pyr-NH2.28 Moreover, a peak attributed to nitrogen appeared at around 400 eV, due to its presence in the fullerene derivative (Fig. S2, ESI†).
Raman spectroscopy provided further proof of the FG reduction due to the nucleophilic substitution and the formation of the G–C60 hybrid (Fig. S3, ESI†). Whereas the precursor GF is Raman inactive,29 the Raman spectrum of the hybrid exhibited the characteristic D and G bands at around 1330 and 1600 cm−1, respectively, demonstrating the construction of a graphene lattice through the formation of sp2 lattice-carbons (G-band) and the conjugation of C60-pyr-NH2 units through the formation of sp3 hybridized carbons (D-band). The intensity ratio (ID/IG) was found to be 1.29, as also observed for other graphene derivatives prepared via the chemistry of FG.19
Thermogravimetric analysis (TGA) was employed under a nitrogen atmosphere to assess the thermal stability and degree of functionalization of the hybrid (Fig. S4, ESI†). The pristine GF is thermally stable below 400 °C and decomposes in the temperature range of 450–650 °C, losing 75% of its weight.29 On the other hand, decomposition of G–C60 started at 175 °C and completed at 600 °C, exhibiting a weight loss of ∼30% of the initial mass. Importantly, C60-pyr-NH2 started decomposing at a lower temperature (155 °C). According to the thermographs of these materials, the higher thermal stability of the fullerene unit on G–C60 can be ascribed to its covalent conjugation with the graphene lattice.
Additionally, the redox properties of G–C60 were assessed in o-DCB and 0.1 M TBAPF6 as electrolyte vs. Fc/Fc+ with the aid of differential pulse voltammetry (Fig. S5, ESI†). The corresponding differential pulse voltammogram is compared with that acquired from C60-pyr-NH2. In short, C60-pyr-NH2 exhibited three reversible reductions at −1.30, −1.67, and −2.22 V.30,31 Concerning G–C60, the reversible reduction at −2.22 V seems to have been shifted to −1.87 V, implying the existence of interactions between both components.31,32 However, the other two reductions are not clearly observed, specifically the reversible peak at −1.67 V appeared broadened.
The morphology of G–C60 was explored by high resolution transmission electron microscopy (HR-TEM) and atomic force microscopy (AFM) (Fig. 3(A)–(D)). A representative HR-TEM image of G–C60 indicated the hybridization of FG with C60-pyr-NH2. According to Fig. 3(A), spherical species, having a diameter of around 0.8 nm, are linked with graphene layers at their edges. The AFM analysis (Fig. 3(B)–(D)) showed that the height of G–C60 was around 3.0 nm, which corresponded well to the thickness of the hybrid material.
 | ||
| Fig. 3 (A) HR-TEM, and (B)–(D) AFM images and height profile of G–C60. | ||
Finally, the electrocatalytic properties of the G–C60 hybrid were explored. In this context, the as-prepared G–C60 was investigated as an electrocatalyst for the HER via the performance of LSV measurements in a N2-saturated aqueous solution 0.1 M KOH electrolyte. The polarization curves of the synthesized G–C60, precursors GF and C60-pyr-NH2 as well as commercially available Pt/C (20 wt% on carbon black) are illustrated in Fig. 4(A) and summarized in Table S2 in the ESI.† An onset overpotential at −0.562 V versus RHE for G–C60 was recorded, which was more positive than the ones recorded for the precursors C60-pyr-NH2 (at −1.488 V) and GF (at −1.613 V), respectively. Concerning the evaluation at −10 mA cm−2, which is the functional current density required for sufficient hydrogen production, the overpotential of G–C60 was found to be −0.939 V versus RHE, 880 and 914 mV more positive than the recorded ones for C60-pyr-NH2 and GF, respectively. All of the above observations show that G–C60 exhibits much higher electrocatalytic activity for the HER in comparison with the used precursors for its synthesis. The great catalytic performance of the G–C60 hybrid in alkaline media is attributed to the synergistic effects between the materials that promote the intermolecular electron transfer from the graphene nanosheets to the electron-accepting fullerenes;31,32 hence, the negative charge density on the fullerene cages facilitates the proton adsorption step and the overall HER activity.10,13
 | ||
Fig. 4 (A) Polarization curves for the HER of benchmark Pt/C (black line), GF (grey), C60-pyr-NH2 (blue line), and G–C60 before (red line) and after 1000 (dashed red line), 5000 (dashed purple line) and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (dashed orange line) cycles respectively, (B) Tafel slopes, and (C) Nyquist plots of GF (grey), C60-pyr-NH2 (blue) and G–C60 (red), and (D) chronoamperometric response of G–C60 (red), at an applied potential of −1.86 V. 000 (dashed orange line) cycles respectively, (B) Tafel slopes, and (C) Nyquist plots of GF (grey), C60-pyr-NH2 (blue) and G–C60 (red), and (D) chronoamperometric response of G–C60 (red), at an applied potential of −1.86 V. | ||
Insight into the HER kinetics was obtained by the Tafel slopes extracted from the LSV curves and the EIS measurements. The Tafel slope value for G–C60 was found to be 347 mV dec−1, which was lower in comparison with the ones for pristine materials GF and C60-pyr-NH2, 438 and 542 mV dec−1, respectively (Fig. 4(B)), indicating the easier HER kinetics for G–C60. The EIS measurements were conducted at −2 mA cm−2, a potential where significant HER current was recorded, and EIS data were fitted to Randles circuit. According to the Nyquist plots, G–C60 displayed the lowest charge resistance value (Rct = 73 Ω), while GF and C60-pyr-NH2 exhibited higher Rct values, 188 and 266 Ω, respectively (Fig. 4(C)). The lower charge transfer resistance for G–C60 than those of individual components is attributed to the formed π-network due to the nucleophilic substitution of amino-modified fullerene moieties onto FGs and the concurrent reductive defluorination. The latter result is also an extra indication for the successful covalent grafting of amino-modified fullerene moieties onto FGs.
Additionally, the long-term stability of G–C60 was evaluated by durability studies. The LSV polarization curves for the HER of G–C60 after 1000, 5000 and 10000 cycles exhibited gradually improved electrocatalytic activity (Fig. 4(A)). Further confirmation of this unusual behavior for G–C60 was received by chronoamperometry measurement, which showed enhanced electrocatalytic performance for G–C60 after 52000 seconds (Fig. 4(D)). The self-improving electrocatalytic activity for materials has been reported in other works as well,33,34 attributed to the activation of more catalytic sites in the hybrids during cycling.
Concerning the estimation of the electrochemically active surface area (ECSA) for G–C60 and its precursors (GF and C60-pyr-NH2), cyclic voltammograms for the HER for each material were measured in a non-faradaic region at scan rates of 50, 100, 200, 300, 400 and 500 mV s−1 (Fig. S6–S8, ESI†). ECSA for G–C60 was calculated at 1.4 cm2, which was higher compared to GF (at 1.0 cm2) and C60-pyr-NH2 (at 0.3 cm2), respectively. These values are in agreement with the electrochemical performances for the HER for each material since the efficient accessibility of the active sites is associated with higher ECSA values.
In summary, we report a non-metal G–C60 hybrid material evaluated as an electrocatalyst for proton reduction displaying improved HER electrocatalytic activity compared to the reference materials FGs and C60–pyr-NH2. The interfacial charge-transfer process between the graphene nanosheets and electron-accepting fullerenes boosted the overall HER activity. This synergistic interaction yielded new active catalytic sites that enhanced the electrocatalytic activity of the material. Notably, the G–C60 electrocatalyst exhibits high durability for up to 52000 seconds. The composition based on light and abundant elements together with the facile one-step synthesis of the hybrid opens new avenues for the efficient covalent integration of carbon allotropes towards the preparation of non-metal all-carbon hybrid materials equipped with electrocatalytic activity.
MO gratefully acknowledges the financial support from the ERC (Consolidator grant 683024 from the Horizon 2020). This work was also supported by the Ministry of Education, Youth and Sports of the Czech Republic (project no. CZ.02.1.01/0.0/0.0/16_019/0000754). Operation of XPS and TEM facilities was partly funded by the Research Infrastructure NanoEnzviCz, supported by the Ministry of Education, Youth and Sports of the Czech Republic (project no. LM2018124). We thank J. Havláková for TGA, M. Petr for XPS and K. Štymplová for Raman spectra measurements.
Conflicts of interest
There are no conflicts to declare.References
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed
.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed
- J. Shi, F. Qiu, W. Yuan, M. Guo and Z.-H. Lu, Chem. Eng. J., 2021, 403, 126312 CrossRef CAS
- R. Paul, L. Zhu, H. Chen, J. Qu and L. Dai, Adv. Mater., 2019, 31, 1806403 CrossRef PubMed
- I. K. Sideri and N. Tagmatarchis, Chem. – Eur. J., 2020, 26, 15397–15415 CrossRef CAS PubMed
- C. Hu, Q. Dai and L. Dai, Cell Rep. Phys. Sci., 2021, 2, 100328 CrossRef CAS
- W. Zhou, J. Jia, J. Lu, L. Yang, D. Hou, G. Li and S. Chen, Nano Energy, 2016, 28, 29–43 CrossRef CAS
- A. Kagkoura, R. Arenal and N. Tagmatarchis, Adv. Funct. Mater., 2021, 31, 2105287 CrossRef CAS
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- M. A. Ahsan, T. He, K. Eid, A. M. Abdullah, M. L. Curry, A. Du, A. R. Puente Santiago, L. Echegoyen and J. C. Noveron, J. Am. Chem. Soc., 2021, 143, 1203–1215 CrossRef CAS PubMed
- M. Chen, R. Guan and S. Yang, Adv. Sci., 2019, 6, 1800941 CrossRef PubMed
- A. R. Puente Santiago, O. Fernandez-Delgado, A. Gomez, M. A. Ahsan and L. Echegoyen, Angew. Chem., Int. Ed., 2021, 60, 122–141 CrossRef CAS PubMed
- R. Gao, Q. Dai, F. Du, D. Yan and L. Dai, J. Am. Chem. Soc., 2019, 141, 11658–11666 CrossRef CAS PubMed
- Z. Du, N. Jannatun, D. Yu, J. Ren, W. Huang and X. Lu, Nanoscale, 2018, 10, 23070–23079 RSC
- J. Guan, X. Chen, T. Wei, F. Liu, S. Wang, Q. Yang, Y. Lu and S. Yang, J. Mater. Chem. A, 2015, 3, 4139–4146 RSC
- A. Hasanzadeh, A. Khataee, M. Zarei and Y. Zhang, Sci. Rep., 2019, 9, 13780 CrossRef PubMed
- D. D. Chronopoulos, A. Bakandritsos, M. Pykal, R. Zbořil and M. Otyepka, Appl. Mater. Today, 2017, 9, 60–70 CrossRef PubMed
- X. Chen, K. Fan, Y. Liu, Y. Li, X. Liu, W. Feng and X. Wang, Adv. Mater., 2022, 34, 2101665 CrossRef CAS PubMed
- D. D. Chronopoulos, M. Medved’, P. Błoński, Z. Nováček, P. Jakubec, O. Tomanec, A. Bakandritsos, V. Novotná, R. Zbořil and M. Otyepka, Chem. Commun., 2019, 55, 1088–1091 RSC
- A. Bakandritsos, M. Pykal, P. Błoński, P. Jakubec, D. D. Chronopoulos, K. Poláková, V. Georgakilas, K. Čépe, O. Tomanec, V. Ranc, A. B. Bourlinos, R. Zbořil and M. Otyepka, ACS Nano, 2017, 11, 2982–2991 CrossRef CAS PubMed
- F. Huang, Y. Li, X. Liu, W. Lai, K. Fan, X. Liu and X. Wang, Chem. Commun., 2021, 57, 351–354 RSC
- Y. Zhang, M. Melchionna, M. Medved, P. Błoński, T. Steklý, A. Bakandritsos, Š. Kment, R. Zbořil, M. Otyepka, P. Fornaserio and A. Naldoni, ChemCatChem, 2021, 13, 4372–4383 CrossRef CAS
- M. F. Sanad, V. S. N. Chava, A. E. Shalan, L. G. Enriquez, T. Zheng, S. Pilla and S. T. Sreenivasan, ACS Appl. Mater. Interfaces, 2021, 13, 40731–40741 CrossRef PubMed
- K. Kordatos, T. Da Ros, S. Bosi, E. Vázquez, M. Bergamin, C. Cusan, F. Pellarini, V. Tomberli, B. Baiti, D. Pantarotto, V. Georgakilas, G. Spalluto and M. Prato, J. Org. Chem., 2001, 66, 4915–4920 CrossRef CAS PubMed
- K. E. Whitener, R. Stine, J. T. Robinson and P. E. Sheehan, J. Phys. Chem. C, 2015, 119, 10507–10512 CrossRef CAS
- D. García, L. Rodríguez-Pérez, M. A. Herranz, D. Peña, E. Guitián, S. Bailey, Q. Al-Galiby, M. Noori, C. J. Lambert, D. Pérez and N. Martín, Chem. Commun., 2016, 52, 6677–6680 RSC
- A. B. Bourlinos, K. Safarova, K. Siskova and R. Zbořil, Carbon, 2012, 50, 1425–1428 CrossRef CAS
- A. Kouloumpis, D. D. Chronopoulos, G. Potsi, M. Pykal, J. Vlček, M. Scheibe and M. Otyepka, Chem. – Eur. J., 2020, 26, 6518–6524 CrossRef CAS PubMed
- J. T. Robinson, J. S. Burgess, C. E. Junkermeier, S. C. Badescu, T. L. Reinecke, F. K. Perkins, M. K. Zalalutdniov, J. W. Baldwin, J. C. Culbertson, P. E. Sheehan and E. S. Snow, Nano Lett., 2010, 10, 3001–3005 CrossRef CAS PubMed
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS
- M. Barrejón, M. Vizuete, M. J. Gómez-Escalonilla, J. L. G. Fierro, I. Berlanga, F. Zamora, G. Abellán, P. Atienzar, J.-F. Nierengarten, H. García and F. Langa, Chem. Commun., 2014, 50, 9053–9055 RSC
- M. R. Cerón, C. Zhan, P. G. Campbell, M. C. Freyman, C. Santoyo, L. Echegoyen, B. C. Wood, J. Biener, T. A. Pham and M. M. Biener, ACS Appl. Mater. Interfaces, 2019, 11, 28818–28822 CrossRef PubMed
- M.-R. Gao, J.-X. Liang, Y.-R. Zheng, Y.-F. Xu, J. Jiang, Q. Gao, J. Li and S.-H. Yu, Nat. Commun., 2015, 6, 5982 CrossRef CAS PubMed
- M. Aygün, M. Guillen-Soler, J. M. Vila-Fungueiriño, A. Kurtoglu, T. W. Chamberlain, A. N. Khlobystov and M. Carmen Gimenez-Lopez, ChemSusChem, 2021, 14, 4973–4984 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc02272a |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |