
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeutral dicyanidoferrate(II) metalloligands for the rational design of dysprosium(III) single-molecule magnets†
Michal
Liberka
 ,
Mikolaj
Zychowicz
,
Wiktor
Zychowicz
and
Szymon
Chorazy
*
,
Mikolaj
Zychowicz
,
Wiktor
Zychowicz
and
Szymon
Chorazy
*
Faculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Krakow, Poland. E-mail: chorazy@chemia.uj.edu.pl
First published on 4th May 2022
Abstract
Diamagnetic cis-dicyanidoferrate(II) complexes bearing blocking aromatic diimines, cis-[FeII(CN)2(LNN)2]0 (LNN = 2,2′-bipyridine, 1,10′-phenanthroline) serve as metalloligands to DyIII centres leading to a rigid cyanido-bridged chain of vertex-sharing {DyIII2FeII2} squares which constrains the equatorial plane of embedded 4f metal ions. This results in a novel convenient route to rationally designed single-molecule magnets as the magnetic anisotropy of DyIII centres can be efficiently generated by inserting aromatic N-oxide ligands on labile axial positions.
Magnetic materials play a vital role in our life due to the applications in data storage, electric motors, power generation and conversion, refrigeration, and medical treatment.1 In this context, increasing interest is given to molecular nanomagnets called Single-Molecule Magnets (SMMs) that reveal the slow magnetic relaxation resulting in a hysteresis loop of molecular origin.2 Thus, they should revolutionize the fields of data storage,3 spintronics,4 and quantum computing.5
Lanthanide (Ln) ions arouse primary interest in the field of SMMs as they show strong magnetic anisotropy being the result of spin–orbit coupling and crystal-field effects.6 They offer further functionalization of SMMs due to the efficient luminescence and softness of the crystal lattice.7 The design principles for maximized anisotropy of 4f metal ions are related to the type of the f-electron density distribution for the preferred highest mJ state.8 Oblate-type ions, e.g. Dy3+, need the axial alignment of charged ligands to minimize the energy of mJ = J state while prolate-type ions, e.g. Er3+, require the equatorial alignment of charged ligands.9 These rules, often supported by high complex symmetry,2a,8b are used for the design of Ln-SMMs. Among them, the most impressive results were found for DyIII centres exhibiting huge anisotropy in sandwich-like organometallic complexes.10 Their disadvantage lies in weak stability to chemical stimuli which is important for processing into devices.3 This can be overcome using strategies based on coordination chemistry. The equatorial plane of the DyIII centre can be constrained by weakly coordinating ligands enabling the introduction of ligands with charged donor atoms to the axial positions.2c,8b Besides the application of multidentate ligands controlling the whole coordination sphere,11 this strategy was realized by two main routes. The first one employs solvent molecules occupying the equatorial plane and expanded organic P-/N-/alkyl-/aryl-oxides placed on the axial positions.9a,12 The second one explores macrocyclic ligands constraining the equatorial plane which leaves the axial positions for oxide-type ligands.13 In this regard, there is a continuous need for novel pathways towards Dy-SMMs exploring the axially and equatorially positioned ligands. We have undertaken this challenge exploiting cyanido complexes of transition metals which are efficient metalloligands for Ln-SMMs with luminescent, SHG-active, or proton-conductive properties.14 Cyanido complexes used alone can induce moderate SMM property due to long LnIII-N(cyanido) bond lengths. However, they can provide a rigid backbone for the f- or d-block metal counter-ions,15 which can be used for constraining the equatorial plane of the DyIII coordination sphere. To achieve this, we screened polycyanidometallates looking for a candidate with a sufficient number of cyanido ligands for bridging to 4f metal ion, being also suitably expanded to block the equatorial plane of Ln3+ ions, leaving space for axially aligned ligands. We found that these prerequisites are provided by cis-dicyanidoferrate(II) complexes bearing aromatic diimines, [FeII(CN)2(LNN)2]0 (LNN = 2,2′-bipyridine = bpy, 1,10′-phenanthroline = phen).16 Here, we report four bimetallic coordination chains, {DyIII(MeOH)4[FeII(CN)2(LNN)2]2}(CF3SO3)3·nMeOH (1, LNN = bpy, n = 1; 2, LNN = phen, n = 5) and {DyIII(pyNO)2(MeOH)2[FeII(CN)2 (LNN)2]2}(CF3SO3)3·nMeOH (3, LNN = bpy, n = 0.5; 4, LNN = phen, n = 1.5). They are built of {DyIII2FeII2} squares that constrain the equatorial plane of DyIII centres opening the way to generate the SMM behaviour by the insertion of pyridine N-oxide (pyNO) ligands on the axial positions of DyIII which was investigated using X-ray diffraction data and dc/ac magnetic characteristics analysed by a newly developed relACs programme supported by the ab initio calculations.
Crystals of 1 and 2 were obtained by the self-assembly of DyIII(CF3SO3)3 with cis-[FeII(CN)2(bpy/phen)2] in the MeOH/MeCN solution. Their basic characterization using CHNS analysis, IR spectra and TGA (see Experimental section and Fig. S1 and S2, ESI†) was followed by a single-crystal X-ray diffraction analysis (Fig. 1 and Fig. S3–S5, Tables S1–S6, S11–S15, ESI†). 1 and 2 crystallize in the C2/c space group (Tables S1 and S2, ESI†) and are composed of cyanido-bridged chains of vertex-sharing {DyIII2FeII2} squares crystallizing with CF3SO3− ions and MeOH of crystallization (Fig. S3–S5, ESI†). Each DyIII centre coordinates four CN− ligands from four neighbouring [FeII(CN)2(LNN)2] complexes, while each FeII site links two different 4f metal ions (Fig. 1). As the convergent cis-dicyanido complexes were used, this results in the {DyIII2FeII2} squares sharing the DyIII centres. The coordination sphere of 4f metal centres is completed by MeOH ligands, lying above and below the equatorial plane of Dy–NC–Fe linkages. The DyIII centre and surrounding FeII sites do not lie in the ideal plane as the neighbouring squares are twisted (Fig. S3–S5 and Table S11, ESI†). The DyIII centres are dodecahedral in 2 or of a mixed geometry between dodecahedral and square antiprismatic in 1 (Tables S3–S6 and S12–S15, ESI†). The Dy–N(CN−) and Dy–O(MeOH) distances vary in the range of 2.39–2.47 Å in 1 and 2 (Tables S3–S6, ESI†). Each FeII site coordinates two diimine ligands that protrude from a coordination skeleton stabilizing the structure by interchain π–π stacking (Fig. S8, ESI†). The addition of pyNO to the same solution as used for 1 and 2 gives the crystals of 3 and 4, respectively (see Experimental section). 3 and 4 crystallize in the Pccn space group but they are similar to 1 and 2 being built of CN−-bridged chains of {DyIII2FeII2} squares crystallizing with CF3SO3− ions and MeOH molecules (Fig. 1 and Fig. S3, S6, S7, Tables S1–S2, S7–S15, ESI†). The difference lies in pyNO ligands attached to the DyIII centres. Each 4f metal ion coordinates two pyNO ligands replacing two out of four MeOH ligands present in the DyIII complexes in 1 and 2. They are located on the axial positions of square-antiprismatic DyIII centres, above and below the equatorial plane given by four Dy–NC–Fe linkages. The Dy–O(pyNO) distances are shorter (ca. 2.36 Å) than Dy–O(MeOH) and Dy–N(CN−) ones (2.42–2.45 Å, Tables S7–S10, ESI†). However, its vertical alignment is not perfectly axial as depicted by the O(pyNO)–Dy–O(pyNO) angle, 124.6(2)° (3) and 127.9(3)° (4), which is related to the twist between neighbouring {DyIII2FeII2} squares. The alignment of pyNO ligands is controlled by intrachain π–π stacking with the LNN ligands which are also involved in interchain π–π interactions (Fig. S6–S8, ESI†). The validity of the structural models were confirmed by the P-XRD (Fig. S9, ESI†).
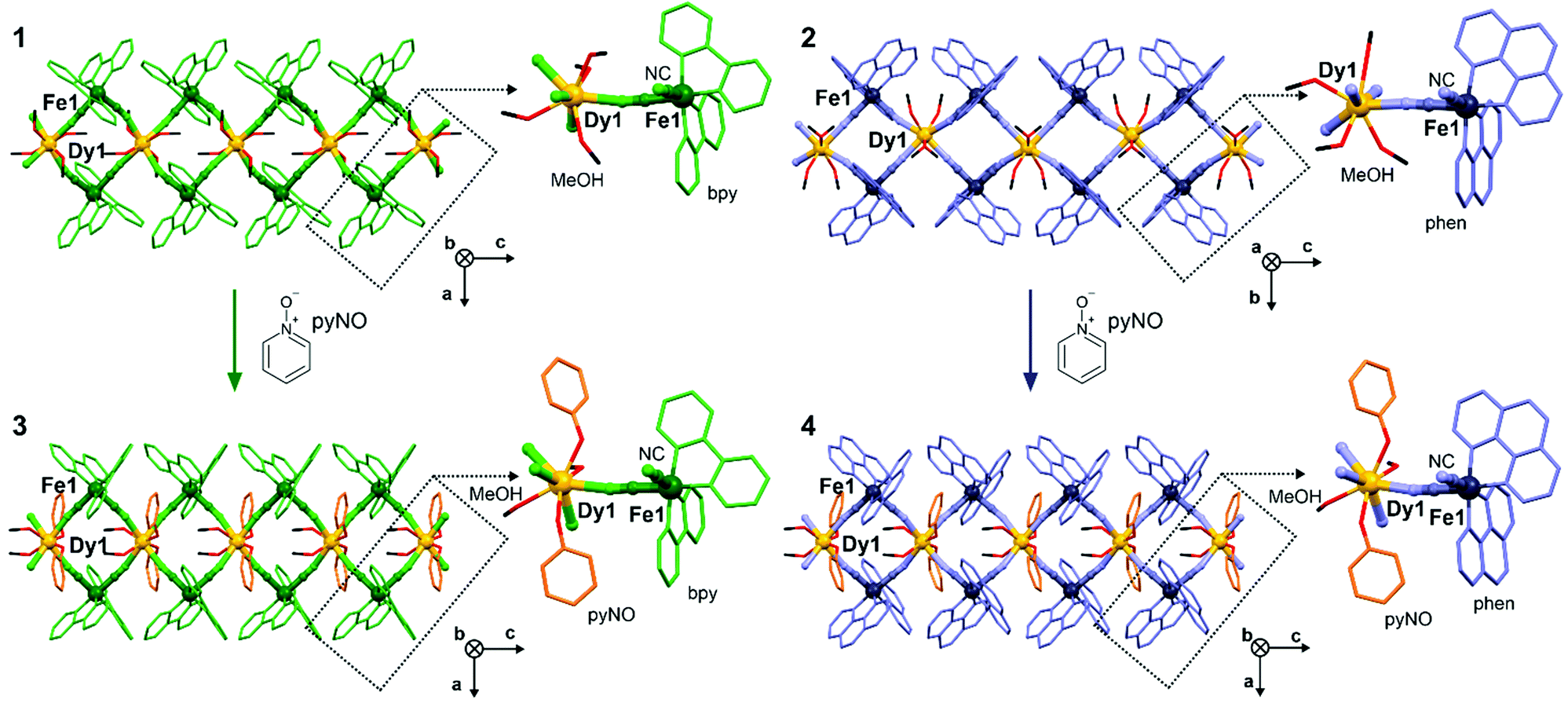 | ||
| Fig. 1 The views of cyanido-bridged chains of vertex-sharing {DyIII2FeII2} squares of 1–4 shown together with respective molecular building units. | ||
The direct-current (dc) magnetism for 1–4 is similar (Fig. S10, ESI†), including the decrease of the χMT product upon cooling related to the thermal depopulation of higher-lying mJ states of the ground DyIII multiplet as well as the featureless course of the M(H) plots at 1.8 K. These properties can be assigned to the single-ion effects of the DyIII complexes which are magnetically isolated (J of ca. –0.07 cm−1 as determined for 3, see Fig. S10, ESI†) due to their separation in the crystal lattice by diamagnetic FeII centres.16 The alternate-current (ac) magnetism of 1–4 is shown in Fig. 2 and Fig. S11–S22, Tables S16–S18 (ESI†). Under zero-dc-field, 1 and 2 do not show a distinct slow magnetic relaxation effect as only the onsets of χM” susceptibility were detected in the high-frequency region (Fig. 2a and b, the insets). However, the χM” signals in 1 and 2 increase and shift to lower frequencies upon the applied dc field (Fig. S11 and S13, ESI†). Under the optimal dc field of 800 Oe, the ac susceptibility was investigated for variable T showing magnetic relaxations below 4 K (1) or 3 K (2) (Fig. S12 and S14, ESI†). Only for 1, the accessible range of relaxation times, determined using the Debye model, enables the analysis of overall relaxation by taking into account three processes: direct, Raman, and QTM (Fig. S11–S22, ESI†). The best-fit parameters prove that the H-induced magnetic relaxation in 1 is dominated by the Raman process in the 2–4 K range while by the QTM below 2 K (Fig. S12 and Tables S17, S18, ESI†). On the contrary, compounds 3 and 4 exhibit distinct maxima on the χM”(ν) plots under zero-dc-field (Fig. 2a and b). They appear almost at the same frequency in the 2–4 K range due to the strong QTM. Then, they shift to higher frequencies being detectable to 10 K in the 1–1000 Hz range. The full set of ac magnetic data for 3 and 4, including field-dependences at 5 K as well as T-dependences at zero and optimal (1 kOe) dc fields, was fitted using the Debye model (Fig. S15–S20, ESI†). The extracted relaxation times were analysed using the simultaneous, 3-D fitting based on all obtained curves, τ(T, H), taking into account direct, Raman, and QTM processes (Fig. 2c, d and Fig. S21, ESI†). All analyses were realized by a purposefully developed relACs programme that enables the fitting of ac data to various models and further 3-D fitting of τ(T, H) curves with the visualization of contributions from relaxation processes which was shown for 3–4 in Fig. 2c and d (for details see ESI,† Fig. S23). We also performed an alternative fit taking into account the Orbach relaxation with a fixed energy barrier, ΔE, taken from the ab initio calculations (Fig. S22, ESI†). The obtained results are consistent with the former fitting, which confirms the major role of the Raman process. The best-fit parameters reproduce well the experiment indicating that the energy barriers of Orbach relaxation of 80.3 and 85.9 cm−1 for 3 and 4, respectively, can be postulated (Fig. S15–S22 and Table S16, ESI†). Despite these values, magnetic relaxation is observed only up to 10 K which is due to the Raman relaxation becoming the main pathway above 7 or 3 K for zero and optimal dc fields, respectively (Fig. 2c, d and Table S17, ESI†). Slightly better SMM features were found for 4 which is due to the larger O(pyNO)–Dy–O(pyNO) angle and the shorter Dy–O(pyNO) distance increasing the ΔE, contributing to quenching of other relaxation routes, e.g. QTM.
 | ||
| Fig. 2 Alternate-current (ac) magnetism of 1–4: the frequency dependences of out-of-phase susceptibility, χ′′, for the 2–10 K range at Hdc = 0 Oe for 3 (a) and 4 (b), together with the fitting to the Debye model (solid lines), compared with the ac signals at Hdc = 0 Oe for 1 and 2 (the insets), and the T-dependences of relaxation time at Hdc = 0 Oe and Hdc = 1000 Oe for 3 (c) and 4 (d). In c and d, circle points show the experimental data, solid lines show the best-fit curves obtained taking into account the combined contribution from direct, Raman, and QTM processes while dashed lines show the separate contributions from each process. | ||
Insight into the transition from weak to distinct SMM features from 1–2 to 3–4 is given by ab initio calculations of a CASSCF/RASSI/SINGLE_ANISO type (Fig. 3 and Tables S19–S21, Fig. S24, and the comment in ESI†). They show that the ground DyIII multiplets in 1–2 consist of the mixture of |J, mJ〉 states of |15/2, ± 15/2〉 (main contribution) and |15/2, ± 11/2〉 (Table S20 and S21, ESI†). The significant mixing for the ground states in 1–2 explains the strong QTM effect and the lack of field-free slow magnetic relaxation. However, the SMM effect appears in 1–2 under a dc field, and, for 1, the energy of the first excited mJ level of 60.0 cm−1 can serve as the estimation of the ΔE of Orbach relaxation, considered in the broadened fit to the ac data (as for 3–4, see Table S16, ESI† and above). The insertion of pyNO in 3–4 improves the magnetic axiality as the related ground DyIII multiplets consist of almost pure |15/2, ± 15/2〉 states with the admixtures of other states below 0.8% (Tables S20 and S21, ESI†). This gives the gz values > 19.8, higher than for 1–2, but non-negligible transversal components remain. It explains the observation of field-free SMM behaviour with the not fully quenched QTM. The energies of the first excited mJ levels in 3–4 can be connected with the Orbach energy barriers; however, it is not crucial as the Raman relaxation dominates. The dramatic changes in anisotropy in 3–4 are proved by the modified alignment of magnetic easy axes (Fig. 3 and Fig. S24, ESI†). In 1–2, they are arranged in the plane formed by four cyanido bridges which provide the most negative charges. In 3–4, the easy axes deflect toward the direction of pyNO ligands proving that they are responsible for enhanced SMM features.
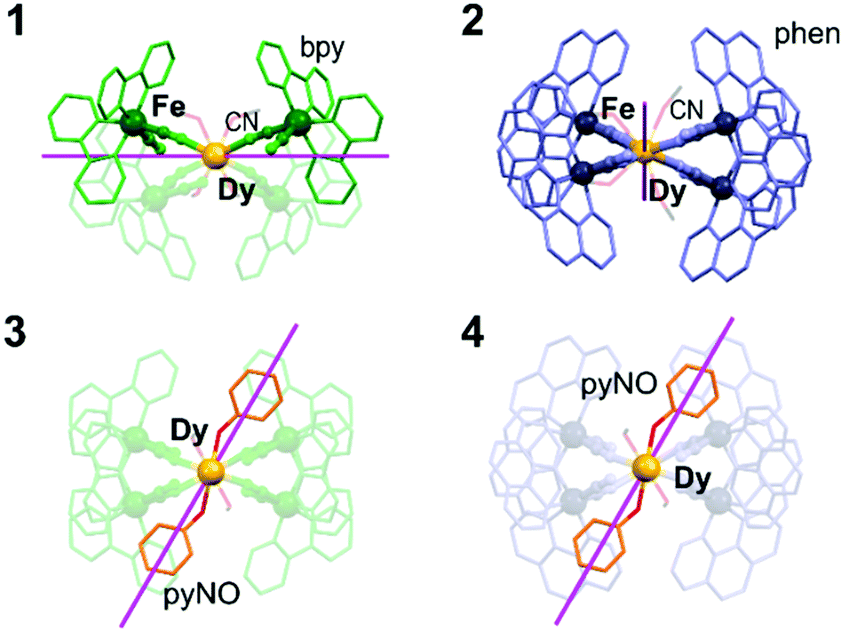 | ||
| Fig. 3 The alignment of easy magnetic axes of Dy complexes in 1–4, determined by the ab initio method (Tables S20 and S21, ESI†). | ||
We report a convenient strategy for the design of DyIII SMMs based on the application of cis-dicyanidoferrate(II) complexes blocked by diimine ligands. Despite their overall zero charges, unusual among cyanidometallates,14 they serve as metalloligands for Dy3+ ions leading to the rigid chain structure that constrains the equatorial plane of the 4f metal centre. As a result, to the axial positions of the DyIII complex, primarily occupied by solvent molecules, one can introduce the organic-oxide-type ligands generating SMM behaviour. This strategy is an alternative to typical approaches using weakly coordinated solvents or macrocyclic ligands occupying the equatorial plane of the DyIII centres.12,13 As ligands on both metal sites can be modified, or the FeII centres can be replaced with other d-block metals,17 this approach opens the route to Dy-SMMs revealing optimized magnetic anisotropy, e.g. by amending the coordination geometry or the axial ligand field,12 and further functionalized towards luminescent and chiro-optical properties. We will search for more rigid cyanido-bridged skeletons leaving two axially positioned solvent molecules for incorporation of charged organic ligands.
This work was financed by the National Science Centre, Poland the OPUS-15 project no. 2018/29/B/ST5/00337. M. Z. acknowledges the Polish Ministry of Science and Higher Education for the Diamond Grant (DI2018 017948).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) O. Gutfleisch, M. A. Willard, E. Brück, C. H. Chen, S. G. Sankar and J. P. Liu, Adv. Mater., 2011, 23, 821 CrossRef CAS PubMed; (b) A. V. Kimel and M. Li, Nat. Rev. Mater., 2019, 4, 189 CrossRef.
- (a) N. Ishikawa, M. Sugita, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694 CrossRef CAS PubMed; (b) S. K. Gupta and R. Murugavel, Chem. Commun., 2018, 54, 3685 RSC; (c) F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400 CrossRef CAS PubMed.
- M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, E. Sciancelepore, A. M. Talarico, M.-A. Arrio, A. Cornia and D. Gatteschi, Nat. Mater., 2009, 8, 194 CrossRef CAS PubMed.
- A. Candini, S. Klyatskaya, M. Ruben, W. Wernsdorfer and M. Affronte, Nano Lett., 2011, 11, 2634 CrossRef CAS PubMed.
- A. Gaita-Arino, F. Luis, S. Hill and E. Coronado, Nat. Chem., 2019, 11, 301 CrossRef CAS PubMed.
- (a) D. N. Woodruff, R. E.-P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5510 Search PubMed; (b) P. Zhang, L. Zhang and J. Tang, Dalton Trans., 2015, 44, 3923 RSC; (c) Z. Zhu, M. Guo, X.-L. Li and J. Tang, Coord. Chem. Rev., 2019, 378, 350 CrossRef CAS; (d) Z. Zhu, X.-L. Li, S. Liu and J. Tang, Inorg. Chem. Front., 2020, 7, 3315 RSC.
- (a) F. Pointillart, B. Le Guennic, S. Golhen, O. Cador, O. Maury and L. Ouahab, Chem. Commun., 2013, 49, 615 RSC; (b) Y. Xin, J. Wang, M. Zychowicz, J. J. Zakrzewski, K. Nakabayashi, B. Sieklucka, S. Chorazy and S. Ohkoshi, J. Am. Chem. Soc., 2019, 141, 18211 CrossRef CAS PubMed; (c) J. Wang, J. J. Zakrzewski, M. Zychowicz, V. Vieru, L. F. Chibotaru, K. Nakabayashi, S. Chorazy and S. Ohkoshi, Chem. Sci., 2021, 12, 730 RSC; (d) R. Marin, G. Brunet and M. Murugesu, Angew. Chem., Int. Ed., 2021, 60, 1728 CrossRef CAS PubMed.
- (a) J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078 RSC; (b) J.-L. Liu, Y.-C. Chen and M.-L. Tong, Chem. Soc. Rev., 2018, 47, 2431 RSC.
- (a) Y.-C. Chen, J.-L. Liu, L. Ungur, J. Liu, Q.-W. Li, L.-F. Wang, Z.-P. Ni, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 2829 CrossRef CAS PubMed; (b) P. Zhang, L. Zhang, C. Wang, S. Xue, S.-Y. Lin and J. Tang, J. Am. Chem. Soc., 2014, 136, 4484 CrossRef CAS PubMed; (c) Z. Zhu, Y.-Q. Zhang, X.-L. Li, M. Guo, J. Lu, S. Liu, R. A. Layfield and J. Tong, CCS Chem., 2021, 3, 338 CrossRef.
- B. M. Day, F.-S. Guo and R. A. Layfield, Acc. Chem. Res., 2018, 51, 1880 CrossRef CAS PubMed.
- J. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 5441 CrossRef CAS PubMed.
- (a) S. K. Gupta, T. Rajeshkumar, G. Rajaraman and R. Murugavel, Chem. Sci., 2016, 7, 5181 RSC; (b) Y.-S. Ding, N. F. Chilton, R. E.-P. Winpenny and Y.-Z. Zheng, Angew. Chem., Int. Ed., 2016, 55, 16071 CrossRef CAS PubMed; (c) A. B. Canaj, M. K. Singh, C. Wilson, G. Rajaraman and M. Murrie, Chem. Commun., 2018, 54, 8273 RSC.
- (a) A. B. Canaj, S. Dey, E. R. Marti, C. Wilson, G. Rajaraman and M. Murrie, Angew. Chem., Int. Ed., 2019, 58, 14146 CrossRef CAS PubMed; (b) A. B. Canaj, S. Dey, C. Wilson, O. Cespedes, G. Rajaraman and M. Murrie, Chem. Commun., 2020, 56, 12037 RSC; (c) Z. Zhu, C. Zhao, T. Feng, X. Liu, X. Ying, X.-L. Li, Y.-Q. Zhang and J. Tang, J. Am. Chem. Soc., 2021, 143, 10077 CrossRef CAS PubMed.
- (a) S. Chorazy, J. Wang and S. Ohkoshi, Chem. Commun., 2016, 52, 10795 RSC; (b) M. Andruh, Chem. Commun., 2018, 54, 3559 RSC; (c) J. Wang, J. J. Zakrzewski, M. Heczko, M. Zychowicz, K. Nakagawa, K. Nakabayashi, B. Sieklucka, S. Chorazy and S. Ohkoshi, J. Am. Chem. Soc., 2020, 142, 3970 CrossRef CAS PubMed.
- E. V. Alexandrov, A. V. Virovets, V. A. Blatov and E. V. Peresypkina, Chem. Rev., 2015, 115, 12286 CrossRef CAS PubMed.
- (a) I. Georgieva, A. J.-A. Aquino, N. Trendafilova, P. S. Santos and H. Lischka, Inorg. Chem., 2010, 49, 1634 CrossRef CAS PubMed; (b) M. Liberka, K. Boidachenko, J. J. Zakrzewski, M. Zychowicz, J. Wang, S. Ohkoshi and S. Chorazy, Magnetochemistry, 2021, 7, 79 CrossRef CAS.
- T. Abe and K. Shinozaki, Inorg. Chem., 2005, 44, 849 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, IR spectra, TGA, detailed structural analysis, detailed dc/ac magnetic analyses, details concerning the relACs programme, details of the ab initio calculations. CCDC 2123814 (1), 2123813 (2), 2123815 (3) and 2123812 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc02238a |
| This journal is © The Royal Society of Chemistry 2022 |