
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The combination of laser photodissociation, action spectroscopy, and mass spectrometry to identify and separate isomers
Samuel J. P.
Marlton
 and
Adam J.
Trevitt
*
and
Adam J.
Trevitt
*
Molecular Horizons and School of Chemistry and Molecular Biosciences, University of Wollongong, Wollongong, New South Wales 2522, Australia. E-mail: adamt@uow.edu.au
First published on 22nd July 2022
Abstract
The separation and detection of isomers remains a challenge for many areas of mass spectrometry. This article highlights laser photodissociation and ion mobility strategies that have been recently deployed to meet this challenge with focus on small molecule isomers including protonation isomers, structural isomers, conformation isomers and new studies emerging on chiral isomers. Laser techniques span UV and visible laser photodissociation, time-resolved pump–probe schemes and application of laser hole-burning arrangements to assign isomers within selected ion populations. Also surveyed are applications of ion mobility strategies to separate isomers followed by laser spectroscopic techniques to assign the separated ions. Ultimately, with ongoing refinement in hardware and methods, there are clear pathways forward for laser and mass spectrometry techniques to make decisive breakthroughs in understanding how isomeric details affect biological processes, physiology and disease.
 Samuel J. P. Marlton | Sam Marlton grew up in Kiama, Australia. He completed his BScience (Honours) degree at the University of Wollongong—including a semester at the University of Colorado Boulder—after which he began his PhD in the Laser Chemistry Laboratory at the University of Wollongong under the supervision of Prof. Adam Trevitt. Sam is continuing to pursue his passion for researching excited state properties, gas-phase ion spectroscopy, and quantum chemistry as he is currently working as a research fellow at the University of Melbourne in the group of Prof. Evan Bieske. |
 Adam J. Trevitt | Adam Trevitt completed his PhD at the University of Melbourne (with EJ Bieske, 2006) after studying for BScience (Honours) at the University of Adelaide (2002). He then completed a postdoc at the University of California, Berkeley and LBNL (with SR Leone, 2007–2008) before starting (in 2008) on the academic track at the University of Wollongong (Australia) where he is (since 2020) a Professor of Physical Chemistry. He leads the Laser Chemistry Laboratory where current projects include photoinitiators for LED curing applications, detection and separation of protonation isomers in mass spectrometry and understanding ion-radical chemistry in reactive gas-phase environments. |
Introduction
In mass spectrometry the inability to separate and assign isomers prevents the full extraction of chemical information and this impacts many research areas including metabolomics,1 lipidomics,2 and mass spectrometry imaging.3 Since the basis of mass spectrometry is the measurement of the mass to charge ratio (m/z) of ions, the separation and identification of isomers requires additional strategies that need to be deployed in tandem. Such strategies include ion-mobility,4 collision-induced dissociation,5,6 ion-molecule reactions (e.g. ozone-induced dissociation),7,8 and laser photodissociation techniques.9,10 Over the last few decades, enormous advances in the conformational biomolecule analysis have occurred leveraging on advances in soft-ionisation techniques (e.g. electrospray ionisation (ESI)) together with numerous ion-mobility and computational strategies. Nevertheless, separating and assigning structural isomers for small molecules remains a significant challenge for mass spectrometry and this Feature Article focusses on recent advances in laser photodissociation (PD) mass spectrometry strategies in this context.In the 1970s, some of the first reports emerged which described UV and visible wavelength photodissociation of mass selected ions as a technique to distinguish isomers. Classic examples include the assignment of various isomers of C7H8+ (radical cations of toluene, cycloheptatriene, and norbornadiene) on the basis of distinct PD spectra.11 At the time, lasers generally lacked broadband UV-Vis tunability so these pioneering action spectroscopy experiments deployed arc-lamp/monochromator light sources with ion cyclotron resonance ion trap mass spectrometry.11–13 Studies by the Dunbar and Beauchamp groups are landmark in this area and remain well-cited over 50 years later.11,12,14
Since these pioneering experiments, there have been perhaps two major technical advancements that have enhanced the scope and application of what is often referred to as PD action spectroscopy: (i) ESI/MALDI ionisation methods and (ii) tuneable UV-VIS mid-band OPO laser sources. The impact of ESI/MALDI cannot be easily overstated as these techniques have proliferated across the biological, chemical and physical sciences because they permit the study of an enormous variety of gas phase ions. Across all these fields, there are distinct pre and post ESI/MALDI invention epochs.15–17 The second major advancement is the availability of the solid-state, tuneable, mid-band OPO laser. These are moderately priced (for a table-top laser system) and are becoming increasingly turn-key and (mostly) hands-free operation. The OPO laser systems used in this context are typically pulsed, with repetition rates commonly (but not limited to) 10–100 Hz and pulse-widths commonly ca. 5–10 ns but also picosecond pulse-widths widely available too. The technical advantages of these OPO laser systems are the broadband tunability (spanning hundreds of nanometres) with good pulse energies (mJ), where scanning from the NIR through the visible and into the UV is relatively straightforward. High-energy wavelength cut-offs are typically around 210–225 nm, depending on the system. However, various vendors offer a range of options including deeper UV extensions, higher repetition rates (ca. 1 kHz), high pulse energies (ca. Joules), IR wavelength extensions, compact footprints, “rugged” housings, and more. There are some limitations and downsides to the common OPO laser systems. For example, OPO laser systems have pulse energies that vary significantly over large scanning ranges and the beam output can be rather divergent with irregular beam spot energy distribution. These factors can be problematic when power normalisation is required for scans over large wavelength ranges and laser beam overlap with ions can vary. Nevertheless, OPO laser systems are robust light sources for acquiring UV-Vis PD action spectra of gas-phase ions.
A major portion of the current UV-vis PD action spectroscopy research is conducted with ion traps, both at room temperature and cryogenically cooled. Ion-mobility strategies such as drift tube ion mobility spectrometry (IMS) and field asymmetric ion mobility spectrometry (FAIMS) are also deployed for pre-filtering of ions. In this Feature Article, the advantages of these techniques are discussed. In defining the scope of this perspective, IR PD techniques—such as infrared multiphoton photodissociation (IRMPD)18–25 and infrared photodissociation (IRPD)26–30—and m/z selected photoelectron (PE) spectroscopy31–34 are not included in detail, with the exception of select examples. The interested reader is directed to these aforelisted references. Also, this Feature Article focusses on studies where ESI is the ionisation source. Other methods that generate ions for action spectroscopy include electron impact ionisation35–37 and supersonic expansion followed by photoionisation38 but these areas are not covered. Finally, therefore, this Feature Article is not an extensive review of all PD spectroscopy strategies. For extended resources the reader is directed to the following reviews on ion-mobility,4,39–41 the history of ion spectroscopy,11,13,42 advances and developments in ion spectroscopy,43 UV-vis photodissociation action spectroscopy,44 lasers and mass spectrometry for structure determination,45 conformer resolved photoinduced dynamics for neutral and ionic peptides,46 action spectroscopy of DNA radical cations,47 gas-phase dynamics including isomer selective spectroscopy,48 biological relevant ions with room temperature49–51 and cryogenic ion spectroscopy.52,53
How does UV-vis PD action spectroscopy work?
Typically, UV-vis PD action spectroscopy is a method that measures the yield of photoproduct ions as a function of photon energy arising from the irradiation of an m/z-selected precursor ion population. Although details can vary, a typical procedure is as follows. A target analyte is introduced from solution via ESI, which serves to transfer the analyte from the liquid solution into the gas phase and (if required) to ionise the analyte. Ionisation often arises by protonation (cation) or deprotonation (anion). Charge can also be provided by non-covalent attachment of cations (e.g. Li+, Na+, K+, and NH4+) or anions (e.g. Cl−, I−, and CH3COO−). Alternatively, target ions may be a native cation or anion. At some point following this liberation of the analyte into the gas phase, m/z isolation of the target precursor ions is performed by a quadrupole ion guide or within an ion trap. This is a vital step that removes all other ions and provides an essentially background-free baseline in preparation for the detection of photoproduct ions. The target ions are then irradiated with a laser pulse (or many pulses). Following this, a mass spectrum is recorded and the intensity of any product ions arising from the laser pulse is measured against the total ion count. This sequence may be repeated depending on the desired signal-to-noise ratio. The laser wavelength is then altered, at the desired step size, and the process is repeated. At each laser wavelength, the PD yield is recorded as either the sum of all the photoproduct ions or a single m/z photoproduct ion spectrum can be plotted as a subset. The success of photodissociation action spectra depends on whether the photon is absorbed (the photon energy corresponds to an electronic transition) and secondly that the photon energy is above the dissociation limit to form product ions (or above any rate limiting barriers that might impede dissociation). Fig. 1 shows two examples where action spectra have been reported using 1-photon mediated photodissociation action spectroscopy and a case where a 2-photon strategy can probe the S1 state, which is below the photodissociation limit, and a second photon is required to generate photoproducts. In the latter case, the two-photon process occurs within the same nanosecond laser pulse and the multiphoton process is confirmed by tracking the power dependence, as explained in ref. 54. Individual product ions may have different PD action spectra, which might signify different photodissociation processes for a precursor ion or that there are different populations within the precursor ion population (e.g. isomers). Interpretation of the PD yield should also take into any account background ions (that are not laser dependent) by background (laser on–laser off) subtraction and also importantly any variations in laser output should be taken into account by laser energy normalisation.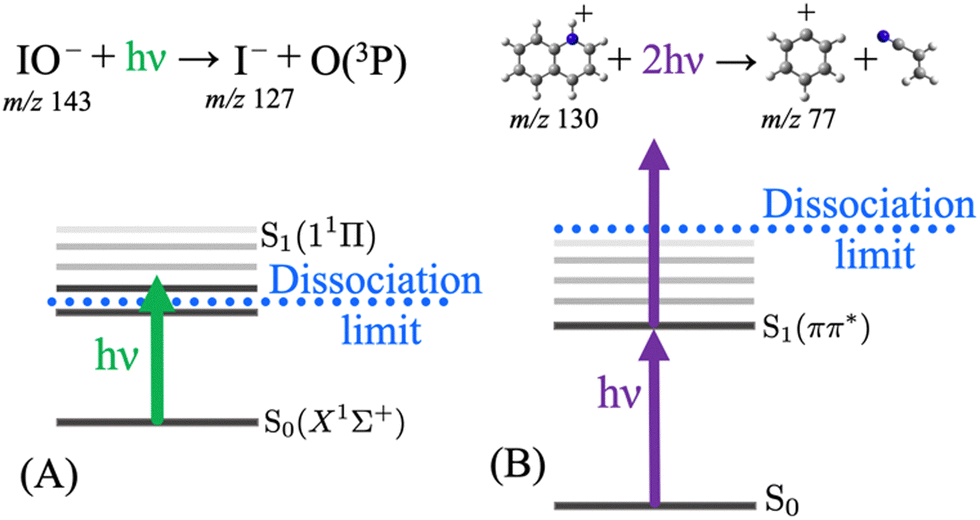 | ||
| Fig. 1 Photodissociation action spectra can be recorded resulting from 1-photon dissociation, for example as depicted for (A) IO− excited into the lowest singlet excited state by one photon (which can be tuned to record the action spectrum). Alternatively, resonant 2-photon mediated photodissociation action spectra are possible as is the case for (B) protonated quinoline excited into the lowest singlet excited state by one photon and the subsequent absorption of a second photon is required to overcome the dissociation limit to form products – tuning the photon provides an action spectrum of the S1 state in this case despite it residing below the photodissociation limit. | ||
The action spectrum can be of different forms. For example, our recent paper on the action spectroscopy of IO− demonstrated how several processes can be investigated following laser irradiation (Fig. 2).55 In this example, IO− is m/z selected and isolated using a room temperature linear ion-trap mass spectrometer and then irradiated. Any loss of IO− parent ions can be recorded with a laser-on vs. laser-off procedure—this is a photodepletion spectrum. Another photoactivated process is bond dissociation, where IO− dissociates into I− + O(3P) and thus a photodissociation spectrum is generated by plotting the yield of I− (m/z 127) normalised to the laser-off signal. While no product ions would normally be expected in the laser-off spectrum, the isolation step in an ion trap mass spectrometry can activate fragile ions and give rise to a background of product ions. Since this example is an anion, another laser activated process is photodetachment of electrons, which is plotted by monitoring the drop in the overall ion count normalised to the laser-off signal. In this example, the competition between photodissociation and photodetachment is plotted out in a bias plot and both are competitive across this photon range. The photodissociation action spectrum, Fig. 2(B), has a superior signal-to-noise ratio, since the I− signal is produced on an essentially zero level of background noise. On comparing this to the depletion signals, Fig. 2 panels A and C, these are inherently affected by more noise since they monitor the attenuation of the parent ion signal. The final note on this series of spectra is that they are all acquired simultaneously, and the photodepletion spectrum is deconvoluted in the photodissociation and photodetachment spectra.
 | ||
| Fig. 2 Various signal pathways in the action spectroscopy of IO−: photodepletion of the IO− parent ion signal (A); the photodissociation yield of I− (B); the electron photodetachment yield from IO− (C); and the bias between photodissociation and photodetachment (D). The superior S/N of the photodissociation spectrum (B) compared to the other spectra is notable. All channels are monitored concomitantly in the ion trap experiment. Reprinted with permission from B. I. McKinnon, S. J. Marlton, B. Ucur, E. J. Bieske, B. L. Poad, S. J. Blanksby and A. J. Trevitt, J. Phys. Chem. Lett., 2021, 12, 11939–11944. Copyright 2021 American Chemical Society.55 | ||
For the acquisition of PD action spectra, various experimental arrangements have been deployed, including adaptations to commercial ion traps,56–60 custom built ion-traps,61 commercial triple-quadrupole MS arrangements,62 cryogenic ion traps,53,63–70 and other designs that involve ion mobility selection71–74 or ion-storage rings.75,76 All these arrangements allow laser access to selected ions to induce photodissociation. Some of these setups include ion mobility stages, which will be discussed in detail later.
As demonstrated in the above example, PD action spectroscopy is an indirect spectroscopic method. For ions, direct gas-phase absorption measurements are generally challenging since gas-phase ions are typically available in a relatively low number density compared to that of solution-phase or dense gas-phase systems. The number of ions that can be generated by ESI—and subsequently selected in the mass spectrometer—can range from tens of millions down to a few tens and this is much lower than the number density in solution phase absorption, which is normally well above 1012 molecules cm−3 (nM). For a gas-phase ion packet if, say, ten photons are absorbed by the sample from a laser pulse made up of trillions of photons, then the attenuation of photon intensity would be difficult to measure. Alternatively, since PD action spectroscopy relies on the detection of photoproduct ions at different m/z values from the selected precursor ions it is essentially a background-free measurement and dissociation events are easily recorded even if only a few product ions are generated for each MS cycle.
Another inherent advantage of PD action spectroscopy using mass spectroscopy is that it introduces an element of target selection of ions by their m/z ratio. As such, background signal and contamination are often eliminated. Of course, this m/z selection alone does not allow for the selection of isomers or isobaric contaminants. The separation and identification of isomers are an active area of research development and a strategy for addressing isomer separation is using ion-mobility techniques to separate isomers as will be discussed later. Another notable method for separation is chromatography, which has been coupled to IRMPD spectroscopy.22,77,78 Although ion mobility techniques are a powerful addition for isomer separation and assignment, spectroscopic techniques alone can provide a detailed characterisation of certain isomers.
Isomer ions
There are many reported studies using UV/vis PD action spectroscopy that interrogate isomeric ions that have been selected before introduction into the mass spectrometer. In these cases, pure samples of structural isomers were available so—provided no isomerisation occurs upon delivery of ions—the spectra from these isomers can be compared. Such studies have targeted substituted pyridines,79–82 azaindoles,83,84 diazabenzenes,85,86 indazole and benzimidazole,87 and diazanaphthalenes,82 as well as anionic systems: deprotonated azaindoles,84 nitrophenolate ions,88–90 molecular-iodide clusters,91 and protonated quinoline and isoquinoline.54,92The case of the structural isomers protonated quinoline and isoquinoline is rather straightforward. The PD action spectra for the S1 ← S0 transitions of quinolineH+ and isoquinolineH+ are shown in Fig. 3. These spectra are constructed by tracking the three major product ions, which are the same for both precursor ions.
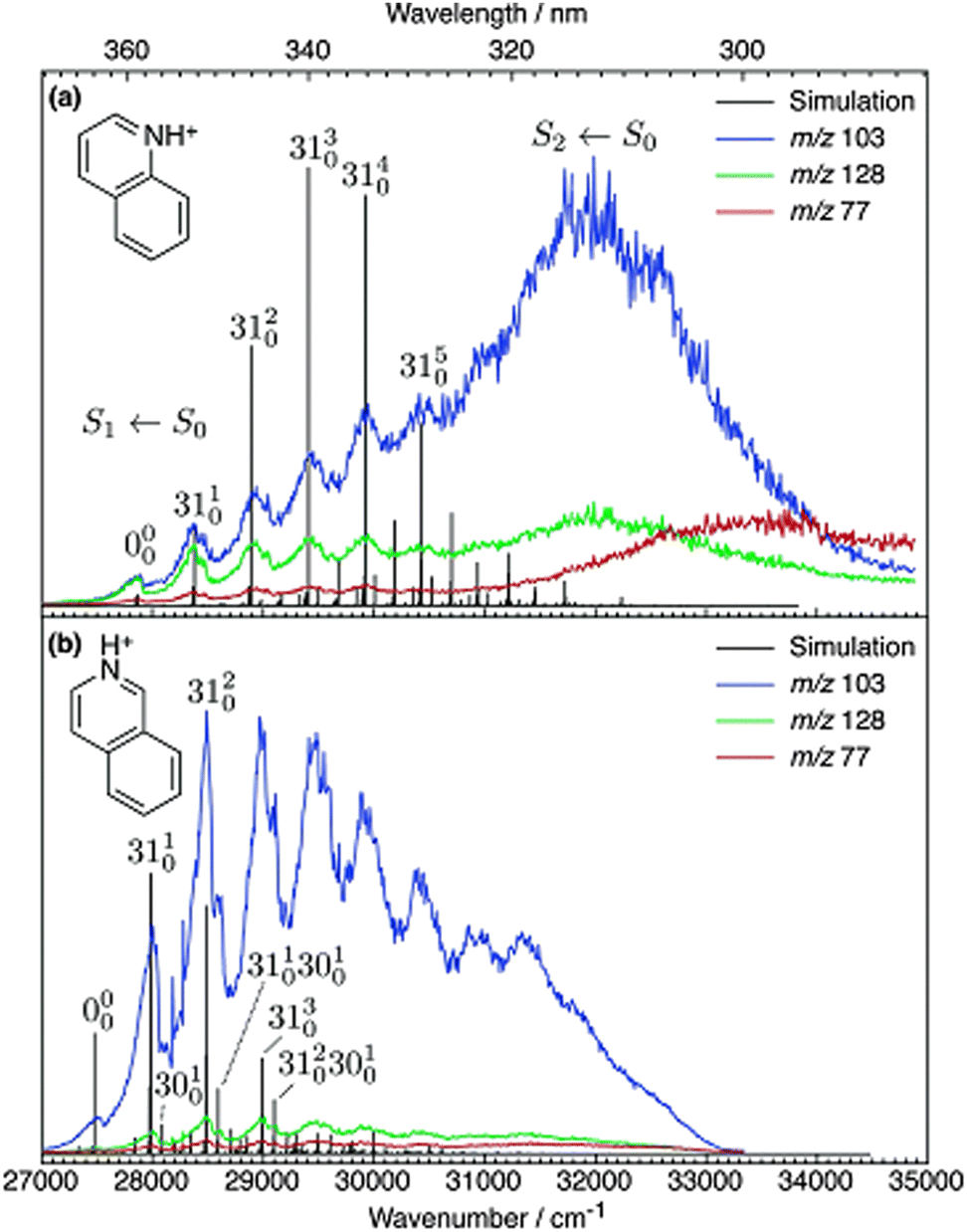 | ||
| Fig. 3 UV PD action spectra for (a) quinolineH+ and (b) isoquinolineH+ ions. Reproduced from ref. 54 with permission from the PCCP Owner Societies.54 These spectra are acquired in a linear ion trap (LTQ). | ||
The pure electronic transition (ν′ = 0 ← ν′′ = 0), labelled 0–0, is often the key value for assigning experimental spectra because it is a clearly defined value in both calculations and experiment.93,94 The difference in the 0–0 energy of protonated isoquinolineH+ and quinolineH+ is ca. 200 cm−1. Note that a hot-band was incorrectly assigned as the 0–0 for isoquinolineH+ in the original publication.54 The 0–0 energies of the two isomers were subsequently measured by Jouvet and co-workers at cryogenic temperatures to be within 200 cm−1.92 The difficulty in characterising small differences between 0–0 energies is compounded by the fact that excited state calculations tend to be less accurate and less robust than ground state calculations. Time dependent density functional theory (TD-DFT) calculations have typical absolute uncertainties in the order of 2000 cm−1 and the coupled cluster method CC2, (which is the gold standard method for excited state calculations of medium sized molecules) has typical uncertainties of ca. 1000 cm−1. Methods like EOM-CCSDT have typical errors of above 200 cm−1 and are too expensive to be routinely used for molecules of this size and larger.95 The fact that EOM-CCSDT and other high level methods—which aspire to reach chemical accuracy (<1 kcal mol−1 or 350 cm−1)—emphasises the importance of benchmark experimental data to validate these claims. In these aforementioned isomers studies, the isomers were generated from chemical standards where the identity of the isomer was known. But what if both isomers were present in one sample? To meet this challenge, Coughlan et al. used the quinolineH+ and isoquinolineH+ reference spectra to assign isoquinolineH+ and quinolineH+ co-sprayed into the instrument and separated within the mass spectrometer using differential ion mobility spectrometry (DMS).96 Therefore, these isomer resolved spectra can serve as references for future diagnostic applications.
A common structural variable that gives rise to isomeric ions in mass spectrometry is variation in the protonation site. Such isomers are termed protomers. Protomers present in the solution may be transferred to the gas phase following ESI, along with protomers that are favoured in the gas phase.21,97–100 However, the relative protomer populations generated under ESI are affected by the ESI conditions and do not necessarily reflect the statistically preferred distributions in either the gas phase or the solution phase.97–100 Because protomer populations are affected by ESI (and because the exact mechanisms by which ions are formed in ESI are not fully understood), it can be difficult to predict which protomer is going to dominate in the experiment. This uncertainty means that the protomers present need to be assigned and characterised.
Room temperature UVPD
Targeting protomers, Matthews and Dessent reported the room temperature UVPD action spectra of protomer isomers co-populated in an ion trap in their study of para-aminobenzoic acid (PABA).9 The room temperature action spectra could be deconvoluted to two protomers of PABA, as shown in Fig. 4. Fig. 4a shows the spectrum of the O-protonated form and Fig. 4b shows the spectrum of the N-protonated form. Because there are major differences in the spectra of these protomer ions, and the major product ions are characteristic to the protomer precursor ions, the isomers are clearly distinguished. Since these product ions are—for the most part—unique to the precursor protomer ion, any isomerization between protomers after photoactivation must be minimal. However, depending on the structure of the molecule, the availability of proton scrambling pathways, and the difference in the protonatable functional groups, protomer ions can give rise to the same photoproduct ions. In this case, the protomers of PABA have different photoproduct ions as well as different action spectra.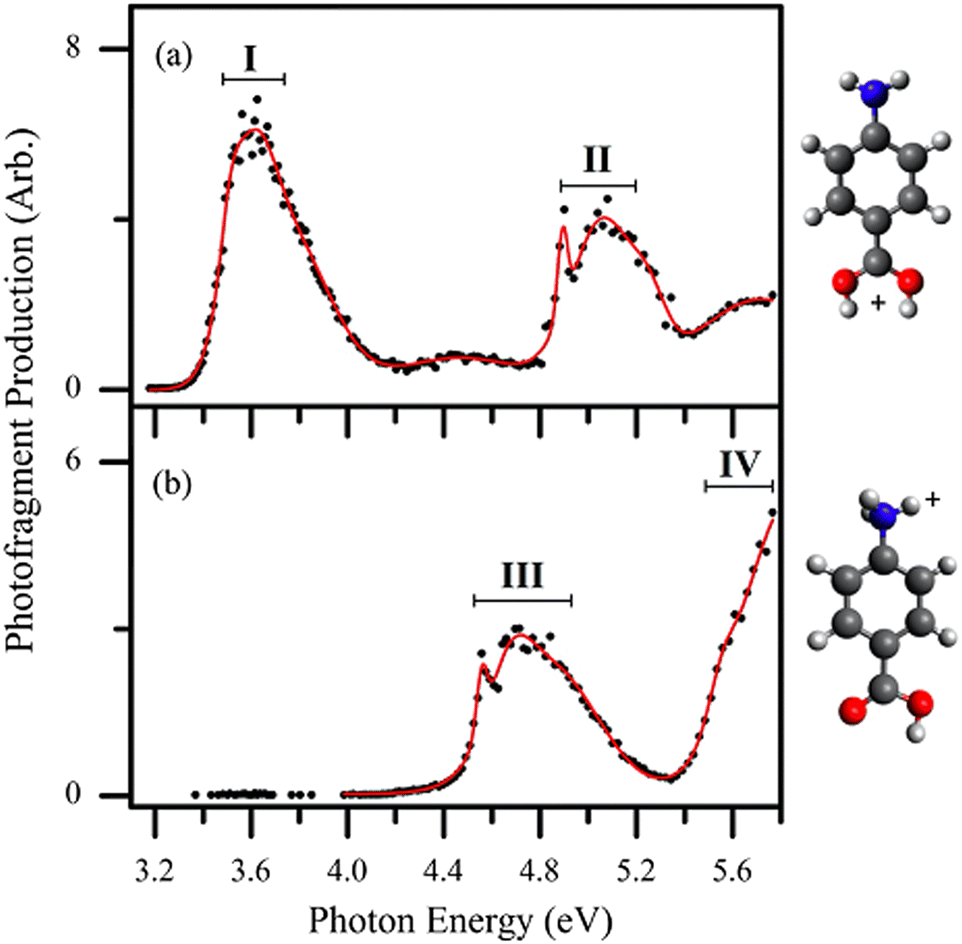 | ||
| Fig. 4 UV PD action spectra for para-aminobenzoic acid (PABA) ions in different solvents following different PD products. The spectra correspond to two protomers co-populated in the ion trap, (a) the O-protomer and the (b) N-protomer, which are distinguishable by their different action spectra and PD products. Reproduced from ref. 9 with permission from the PCCP Owner Societies.9 | ||
Photodissociation product ions can provide insight into assignment of the protomer precursor ion. For example, Fig. 4b follows both the generation of the H atom (−1 Da) and the NH3 fragment (−17 Da), previously assigned to the N protomer.101,102 Therefore, based on the photoproducts, one can begin to assign the spectra to different protomers before analysing each spectrum.
For direct assignment of isomers, photodissociation action spectroscopy can be compared to calculated spectroscopic transitions. For example, the first electronic transition for the O-protonated form of PABA is calculated to be centred around 3.58 eV (using the high level MRCI method) and the peak in the experimental spectrum (Fig. 4a) is centred around 3.51 eV. Similarly, the calculated first transition of N protonated PABA is calculated to be 4.50 eV, which is close to the experimental transition around 4.56 eV (Fig. 4b). This agreement between calculations and experimental spectra—in addition to other observed states—provides an unequivocal assignment of these spectra to the O and N protomers, which are co-located in the ion-trap.
The Dessent group has also deployed PD action spectroscopy to study protomers of flavin ions103 and nicotinamide.104 In these experiments, the various isomers are co-located in the ion trap. Other UV PD studies on nucleobases105,106 distinguished between multiple protomers. These studies demonstrate how the electronic transitions and non-radiative decay are affected by the protonation site, which is noteworthy considering protonation bears some resemblance to the hydrogen bonding and isomerism that occur for nucleobases in DNA. From the PD action spectra of these molecules, their intrinsic photoproperties can be studied. These properties include absorption transitions and non-radiative decay mechanisms. This is benchmark information to understand the effect of more complex environments, including solvent shifts or other environmental factors. UV PD has also been employed to understand how structural changes affect the photochemistry of sunscreens, for example, by distinguishing and characterising how the binding site of a cation can inhibit the excited-state proton transfer that affects the photostability of sunscreen agents.107–109
Nucleobase radical cation isomers,47,110–113 as well as several other biologically relevant radical cation isomers, have been analysed by Turaček and co-workers using UV action spectroscopy in a modified commercial ion trap, which takes advantage of the inert ion trap environment. Reactive radical ions, which would otherwise be transient intermediates that quickly react away, can be isolated and stored for the acquisition of PD spectra.
The Turaček group has also compared the results from the two most common commercial mass spectrometers that are modified for PD action spectroscopy (THERMO LTQ and Bruker AmaZon). These modified setups are shown in Fig. 5a and b.56 The main difference between these arrangements is that for the linear ion trap (THERMO LTQ) optical access is afforded through a window on the rear of the mass spectrometer, whereas optical access is afforded through a window on the top of the mass spectrometer to the 3D ion trap (Bruker AmaZon) with a clear path in and out of the instrument. The spectra in Fig. 2 and 3 were acquired using a THERMO LTQ mass spectrometer, whereas the spectra in Fig. 4, from Dessent and co-workers, were acquired using a Bruker AmaZon mass spectrometer. It is evident that both configurations are suited for PD action spectroscopy; however, these spectra should not be used to directly compare the performance of the two mass spectrometer setups because they also employed different lasers, span different photon energies, and report spectra from different ions. For direct comparisons between equivalent instruments using a THERMO LTQ and a Bruker AmaZon mass spectrometer see ref. 56.
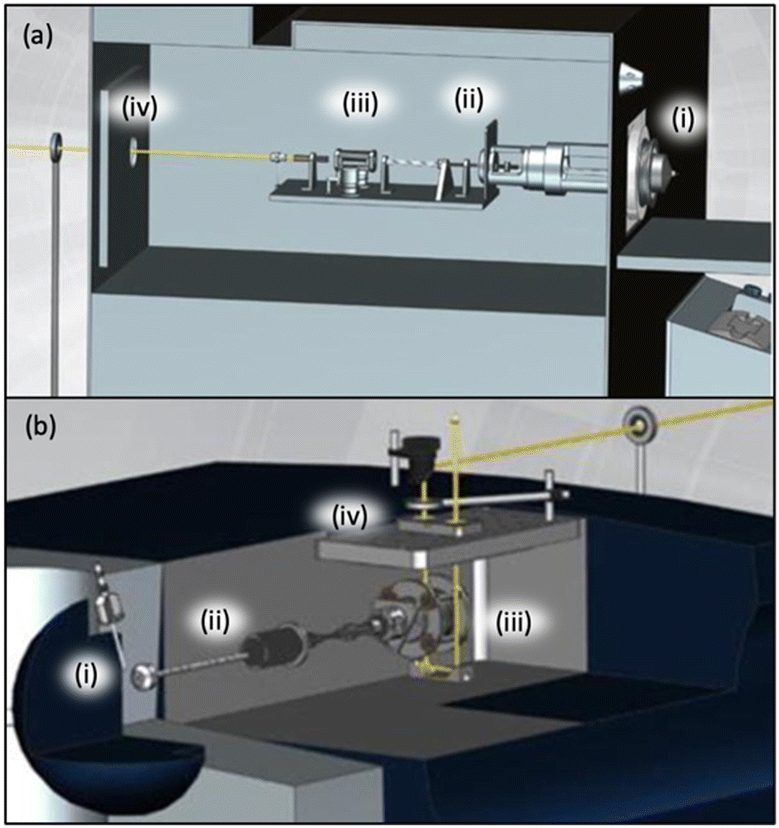 | ||
| Fig. 5 A diagram of how a THERMO LTQ (a) or a Bruker AMAZON mass spectrometer (b) can be adapted to allow optical access to the ion trap is shown. Figure has been amended to include labels of key components indicating ESI ion source region (i), electrostatic ion guides (ii), ion trap (iii), and laser beam (yellow line) entering the mass spectrometer through a window (iv). Adapted from A. Dang, J. A. Korn, J. Gladden, B. Mozzone and F. Tureček, Journal of The American Society for Mass Spectrometry, 2019, 30, 1558–1564 Copyright 2019 American Chemical Society (ref. 56). | ||
Although the are many cases where room temperature PD action spectroscopy can distinguish isomers, there will be many instances where that the room temperature spectra of isomers are inherently too broad and/or congested. Cooling of ions in cryogenically-cooled ion traps can simplify spectra.
Cryogenic UVPD
As is well established, vibronic spectra are generally sharper and better resolved when ions are cooled in cryogenic ion traps as this greatly restricts vibrational state populations and narrows rotational state distributions. PD action spectroscopy of ions confined in cryogenically cooled environments is conducted in a similar manner to the previously described room temperature experiments except that the temperature of the ions is cooled to ca. 3–50 K using a buffer gas, which is typically helium gas. Fig. 6b shows a schematic of a setup employing a cryogenic ion trap as shown in ref. 63.63 The ions are generated by ESI and guided into a 22-pole ion trap, which is cooled by a He cryostat. With colder ions, hot band intensities and rotational broadening are significantly reduced or eliminated and the sharpness of the spectra can be vastly improved.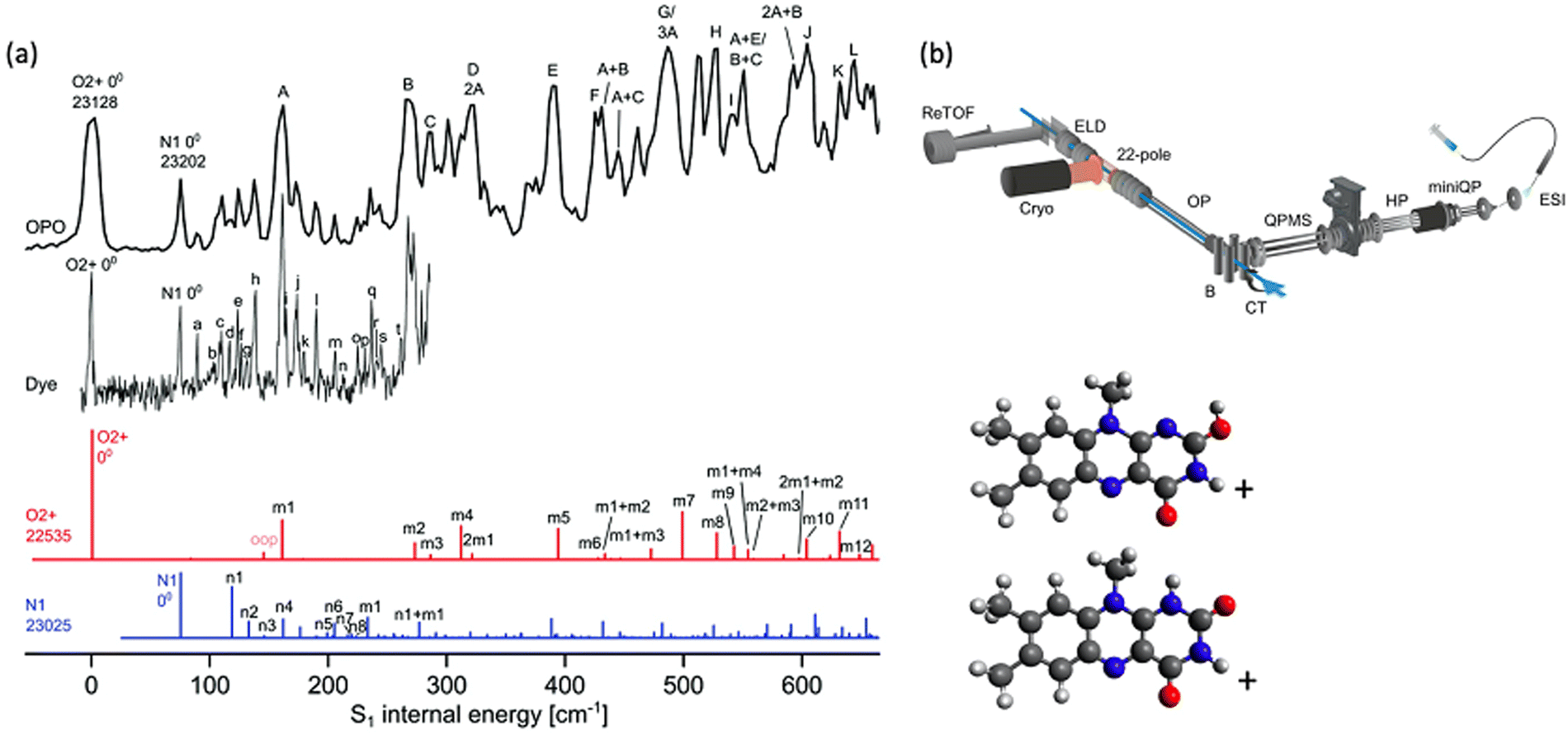 | ||
| Fig. 6 (a) Cryogenic spectra of LFH+ (black) acquired with a mid-band OPO and a sharper-linewidth dye laser. Franck–Condon simulations for two protomers are red and blue. Both protomers are assigned as contributing to the experimental spectrum. Reproduced from ref. 114 with permission from the PCCP Owner Societies.114 (b) A schematic illustration is included to provide a survey of the cryogenic BerlinTrap apparatus used to acquire these spectra. Ions are generated using ESI, accumulated in a miniature quadrupole (miniQP), transferred into a hexapole (HP), m/z selected using a quadrupole mass spectrometer (QPMS), reflected 90° by a quadrupole bender, guided by an octupole (OP) into a cryogenic 22 pole ion trap where they are cooled and can be irradiated by a laser pulse (blue line) after which the ions are detected using a reflectron time of flight mass spectrometer (ReTOF). (b) Reproduced with permission from A. Günther, P. Nieto, D. Müller, A. Sheldrick, D. Gerlich and O. Dopfer, A new cryogenic 22-pole ion trap spectrometer, J. Mol. Spectrosc., 2017, 332, 8–15. Copyright Elsevier, 2017.63 | ||
Cryogenic ion-trap PD action spectroscopy has been used to characterise the protonation or metalation-site isomers for ions of flavins,114–116 nucleobases,117,118 polycyclic aromatic amines,119,120 distinguishing between different spin states of high-spin ions,121,122 and to disentangle E and Z double-bond isomers of hemithioindigo123 and the green fluorescent protein chromophore,124 lipid isomers125 and carbohydrates.126 Similar experiments have measured electron photodetachment (rather than PD) as a function of wavelength for deprotomers of cytosine anions.31
The PD action spectra of protonated lumiflavin (LFH+) are shown in Fig. 6 (from the study by Dopfer and co-workers).114 A lower resolution spectrum is recorded using a mid-band OPO laser (linewidth ca. 4 cm−1) and the high-resolution spectrum is acquired using a dye laser (linewidth ca. 0.014 cm−1). The first major peak (normalised at 0 cm−1) is assigned as the origin of the O protomer. Aided by the simulations, it is apparent that two co-located protomers contribute to the experimental spectrum. The N-protomer 0–0 is assigned at +74 cm−1 from the O-protomer 0–0. Several low frequency excited state vibrational quanta are assigned within a few 100 cm−1 above the 0–0 transition due to both protomers. This vibronic structure allows for the assignment of two protomers based on comparison with FC simulations and calculated 0–0 transition energies. The O2+ protomer (red simulation) is planar in the excited state and is dominated by in-plane vibrations and the geometry of the N1 protomer (blue simulation) is non-planar in the excited state and has a spectrum that has several active low-frequency out of plane vibrations. This illustrates how biological flavins can have altered photoproperties as a consequence of different protonation sites, which then highlights the importance of the flavin environment to biological function. Furthermore, the flavin properties can also be affected by metal cation binding, as other studies using cryogenic PD action spectroscopy have observed that changing the metal binding site can dramatically shift the 0–0 transition energy for the first 1ππ* state of flavin ions.114–116
For protonated lumiflavin, vibronic spectra obtained under cryogenic conditions allow for convincing assignment of protomers. Assignment is achieved by comparison between experimental and calculated results, which typically involves Franck–Condon simulations and calculated 0–0 transition energies.114,115,119,120,127–130 The interplay between experiment and computational chemistry methods is exemplary in this area of research. Accurate calculations are often needed to assign experimental spectra and, in turn, quantum chemical calculations need experimental data for benchmarking and validation.
Natural linewidth-limited peak shapes recorded using cryogenic ion PD spectroscopy can be used to estimate excited-state lifetimes.83,85,118 This is possible because rotational broadening of vibronic peaks should be minimised at low temperatures and generally spectra are much less congested. Using cryogenic ion PD spectroscopy, two protomers of protonated uracil were determined to have excited state lifetimes of 40 fs and 2 ns (where the 2 ns lifetime was measured using a pump–probe).85,118 This difference between lifetimes was attributed to the different energy gaps between the ππ* and nπ* states. Thereby, the effect of lone pairs and protonation site on the photostability of DNA nucleobases was spectroscopically observed.
Pump–probe photodissociation
For co-located isomer ions, if vibronic transitions are sufficiently narrow and separated it is possible to tune a laser to selectively excite a vibronic transition of only one isomer population, leaving the other unaffected. Taking advantage of this, Soorkia et al. selectively excited particular conformers of protonated tyrosine ions using resonant transitions. By then introducing a second (probe) laser pulse, they measured pump–probe PD spectra for various conformers (see Fig. 7).131 A schematic of the experimental setup is also shown in Fig. 7c; all isomers are co-isolated in the ion-trap (Paul trap), which is cooled by a cryostat. Both the pump and probe pulses (see fragmentation laser in Fig. 7c) irradiate the ion cloud in the ion-trap.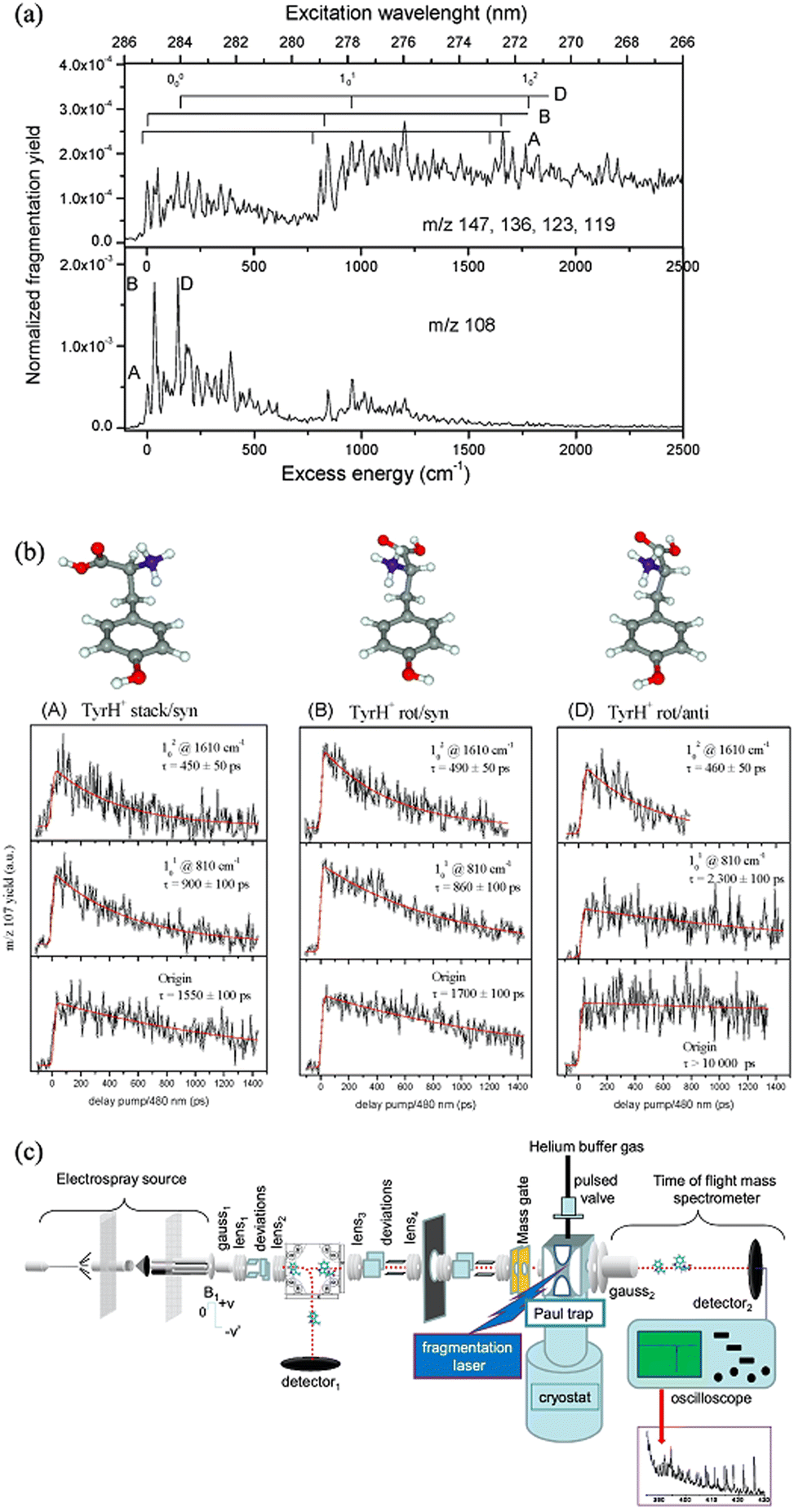 | ||
| Fig. 7 (a) Cryogenic UV PD action spectra of tyrosineH+ ions. (b) pump–probe PD plots following the PD yield of tyrosineH+ ions as a function of time delay between the pump and probe pulses (right). By setting the pump pulse to excite a vibronic peak for only one isomer, each isomer can be selected and its lifetime can be selectively probed. (a and b) Adapted with permission from S. Soorkia, M. Broquier and G. Grégoire, J. Phys. Chem. Lett., 2014, 5, 4349–4355. Copyright 2014 American Chemical Society.131 (c) Schematic of the experimental setup. (c) Adapted with permission from I. Alata, J. Bert, M. Broquier, C. Dedonder, G. Féraud, G. Grégoire, S. Soorkia, E. Marceca and C. Jouvet, J. Phys. Chem. A, 2013, 117, 4420–4427. Copyright 2013 American Chemical Society.64 | ||
This arrangement of the isomer-selective pump–probe experiment was employed to investigate protonated tyrosine isomers131 and nucleobase isomers.117,132 Protonated tyrosine conformers are found to have similar excited state lifetimes at 1610 cm−1 above the 0–0 transition (ca. 450 ps). On the other hand, the 1nπ* lifetimes of protonated cytosine varied by over an order of magnitude and, as previously mentioned, the excited state lifetimes of uracil conformers varied by almost five orders of magnitude.
Other groups have deployed femtosecond or picosecond pump–probe laser configurations combined with photodissociation mass-spectrometry to interrogate excited-state lifetimes of ions in tandem time-of-flight mass spectrometers,133–135 triple-quadrupole mass spectrometers,136 ion storage rings,137,138 custom built ion traps at room temperature,139–141 custom built ion traps that are cryogenically cooled,81,85,132,142 and commercial ion traps.87,143–146 There are also setups using electronic delays for pump–probe photodissociation on the nanosecond to millisecond timescale.117,132,146–151 The excited state dynamics of few gas-phase ions has been investigated experimentally or computationally, especially relative to solution-phase species.
Laser hole burning techniques
A particular isomer population that are co-located with other isomers can be selectively excited by tuning a laser to a characteristic vibronic transition and “burning” out a major fraction of that population. Hole burning spectroscopy takes advantage of this selectivity to obtain a spectrum of only one isomer or conformer. This requires two lasers. First, a single laser photodissociation spectrum would normally be recorded and contain peaks from all isomers/conformers co-located in an ion trap experiment. In the hole-burning experiment, a probe laser is fixed to one peak in the PD spectrum. Then a burn laser is scanned across the spectrum and this is pulsed a short time before (ca. a few milliseconds) the probe laser. Variations in the probe laser PD signal are tracked as the burn laser is scanned. When the burn laser is resonant with a transition from an isomer that is also targeted by the probe laser, a depletion in the probe signal is recorded. For other transitions scanned over by the burn laser, the probe laser signal is unaffected. This process is repeated with a probe laser then shifted to a different peak (Fig. 8 includes several hole-burning spectra). Therefore, the hole-burning strategy provides an isomer-selective depletion spectrum.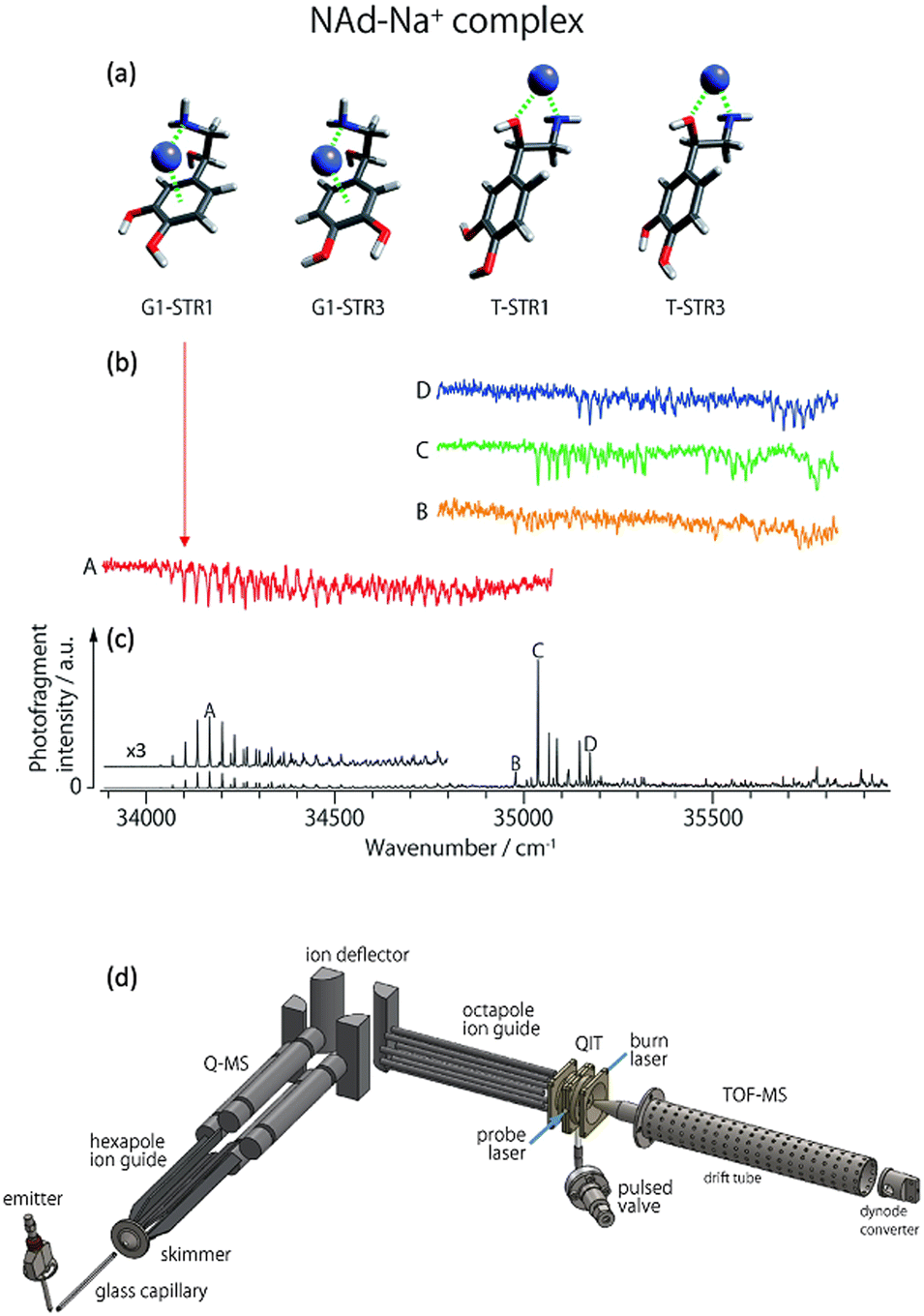 | ||
| Fig. 8 (a) Some low energy isomers of noradrenaline-Na+. (b) Cryogenic UV-PD hole burning spectra of noradrenaline-Na+ isomers. (c) Cryogenic UV-PD spectrum exhibiting the spectrum of all isomers generated by ESI. Figures (a), (b) and (c) reproduced from ref. 152 with permission from the Royal Society of Chemistry.152 By setting a “probe” laser pulse to selectively excite one isomer, the PD from that isomer is detected. By introducing a second laser, an isomer selective spectrum can be generated whereby the spectrum dips when the second laser “burns” out the population of the isomer being selected by the “probe”. (d) A schematic illustration is included to provide a survey of the experimental setup. Ions are generated using ESI, guided through a hexapole, m/z selected using a quadrupole mass spectrometer (Q-MS), reflected 90° by a quadrupole bender, guided by an octupole into a quadrupole ion trap (QIT) where they are cooled and can be irradiated by one or two laser pulses (blue lines) after which the ions are detected using a reflectron time of flight mass spectrometer (TOF-MS). (d) Reproduced with permission from S.-i. Ishiuchi, H. Wako, D. Kato and M. Fujii, High-cooling-efficiency cryogenic quadrupole ion trap and UV-UV hole burning spectroscopy of protonated tyrosine, J. Mol. Spectrosc., 2017, 332, 45–51 Copyright Elsevier, 2017.65 | ||
Examples of hole burning spectra are presented in Fig. 8 where isomer-selective spectra for four noradrenaline-Na+ conformers (red, orange, green and blue traces) are recorded from an experiment with all isomers co-located in the ion trap. The different spectra are acquired by scanning the burn laser and setting the probe laser to selectively excite one of the peaks A, B, C, or D shown in Fig. 8c. A schematic example of the instrumentation for a hole burning experiment is shown in Fig. 8d (reproduced from ref. 65), which incorporates ESI, m/z selection with a quadrupole mass spectrometer (labelled Q-MS). Ions are guided into the ion-trap where they are cryogenically cooled by pulsed helium gas and subsequently irradiated by the burn and probe laser pulses. This hole burning experiment was used to assign the vibronic progression labelled A (red trace) to a closed structure where Na+ interacts with the aromatic π system. This isomer was not observed for the noradrenaline-Li+ ion (not shown), which may explain the different effect that Li+ and Na+ has on the inhibition of noradrenaline molecular recognition.152
Biomolecular ions of considerable structural flexibility like polypeptides and proteins have been analysed using combined cryogenic and IR and UV hole burning and double resonance experiments.30,153–155 For more on cold ion spectroscopy, the interested reader is directed to the following reviews for biological ions by Boyarkin and co-workers52,156 and Soorkia et al.46
Ion mobility spectrometry with photodissociation
Ion mobility spectrometry in its various forms is a method to separate ions by physical properties including size, dipole and buffer–gas affinity. By coupling IMS with PD spectroscopy, isomers can first be separated and then spectroscopically investigated without the complication of co-located isomers.An important example of these techniques used together is from Koyasu et al. targeting isomers of C9+ arising from laser vaporisation of graphite. Various C9+ isomers were separated using drift tube ion mobility spectrometry and then irradiated with 355 nm photons to induce photodissociation (see Fig. 9). In IMS, ions drift through a cell that contains a gas, typically N2 or He. As the ions drift, their mobility is affected by the ion–molecule collisions as quantified by the collision cross section (CCS). Two types of Cn+ isomers are reported in this experiment, linear and cyclic. The linear isomers have a larger CCS and they tumble through the collision cell with longer arrival time delays (peak labelled A in Fig. 9b). The cyclic isomers are more compact with a smaller CCS and, therefore, have the arrival time (peaks labelled B in Fig. 9b).
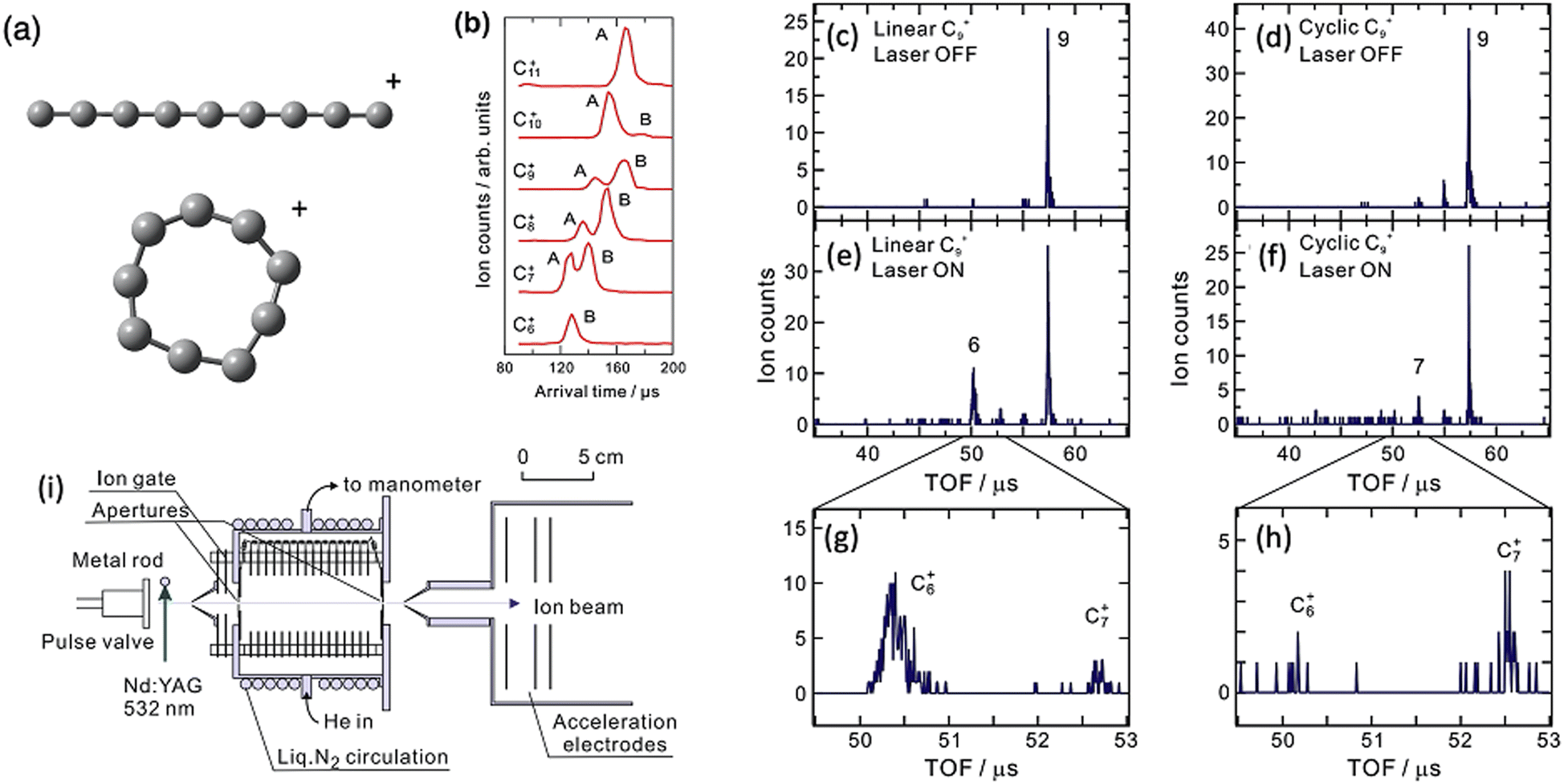 | ||
| Fig. 9 Structures of linear and cyclic C9+ (a), Drift IMS arrival times plots of Cn+ clusters (b), mass spectra of linear C9+ with laser on (c) and laser off (e and g), mass spectrum of cyclic C9+ with laser on (d) and laser off (f and h). Adapted with permission from K. Koyasu, T. Ohtaki, N. Hori and F. Misaizu, Isomer-resolved dissociation of small carbon cluster cations, C7+–C10+, Chem. Phys. Lett., 2012, 523, 54–59 Copyright Elsevier 2012.157 (i) Schematic of experimental setup reproduced from F. Misaizu, N. Hori, H. Tanaka, K. Komatsu, A. Furuya and K. Ohno, Isomer-selected photoreactions of gas-phase cluster ions, Eur. Phys. J. D, 2009, 52, 59–62 with permission of Springer.74 | ||
Because the two C9+ isomers are separated, the ratio between C6+ and C7+ formation is determined, where the linear isomer favours C6+ and the cyclic isomer favours C7+. Because they are separated, the difference is obvious, which is a clear advantage of the combined ion mobility and PD mass spectrometry arrangement. Action spectra are not acquired in this experiment. A subsequent experimental arrangement that combines IMS and cryogenic PD action spectroscopy has presented the electronic spectra of several cyclic carbon cluster ions.158,159
Some other examples of ion mobility coupled with photoelectron spectroscopy or PD using set wavelengths are other carbon cluster ions,160,161 silicon cluster ions,161 trisaccharides,162 DNA anions,163,164 proteins,165–169 flavins,73 and glycans.170 Also, fixed wavelength arrangements have been used for more exotic experiments such as ion-mobility selected gas-phase fluorescence.171–173
Ion mobility spectrometry coupled with spectroscopy
Ion-mobility and PD action spectroscopy deployed together can reveal considerable detail about ions and isomer populations. For example, protomers of benzocaineH+ could be separated first using IMS and then assigned using IRMPD.100Fig. 10b shows that the mobility selected IRMPD spectrum of isomer II matches the calculated spectrum for the N-protomer of benzocaineH+, and the spectrum of I matches the calculated spectrum of the O-protomer. The IRMPD spectra exhibit several diagnostic peaks (like the N-protomer peak at 1250 cm−1 or the O-protomer peak at 1550 cm−1). These peaks in the IRMPD spectra allow for unambiguous assignment of the two ion-mobility separated species. Notably, CID generates the same photoproducts for both protomers and, therefore, does not provide definitive assignment information. However, one could argue that these protomers could be assigned using IRMPD without separation—although the result would be perhaps less definitive without IMS separation. Even though the IMS points to additional isomers (isomers II and II′) the IRMPD spectra of these isomers appear almost identical. The populations of these protomers and isomers can be measured by variation of the ESI solvent (Fig. 10a).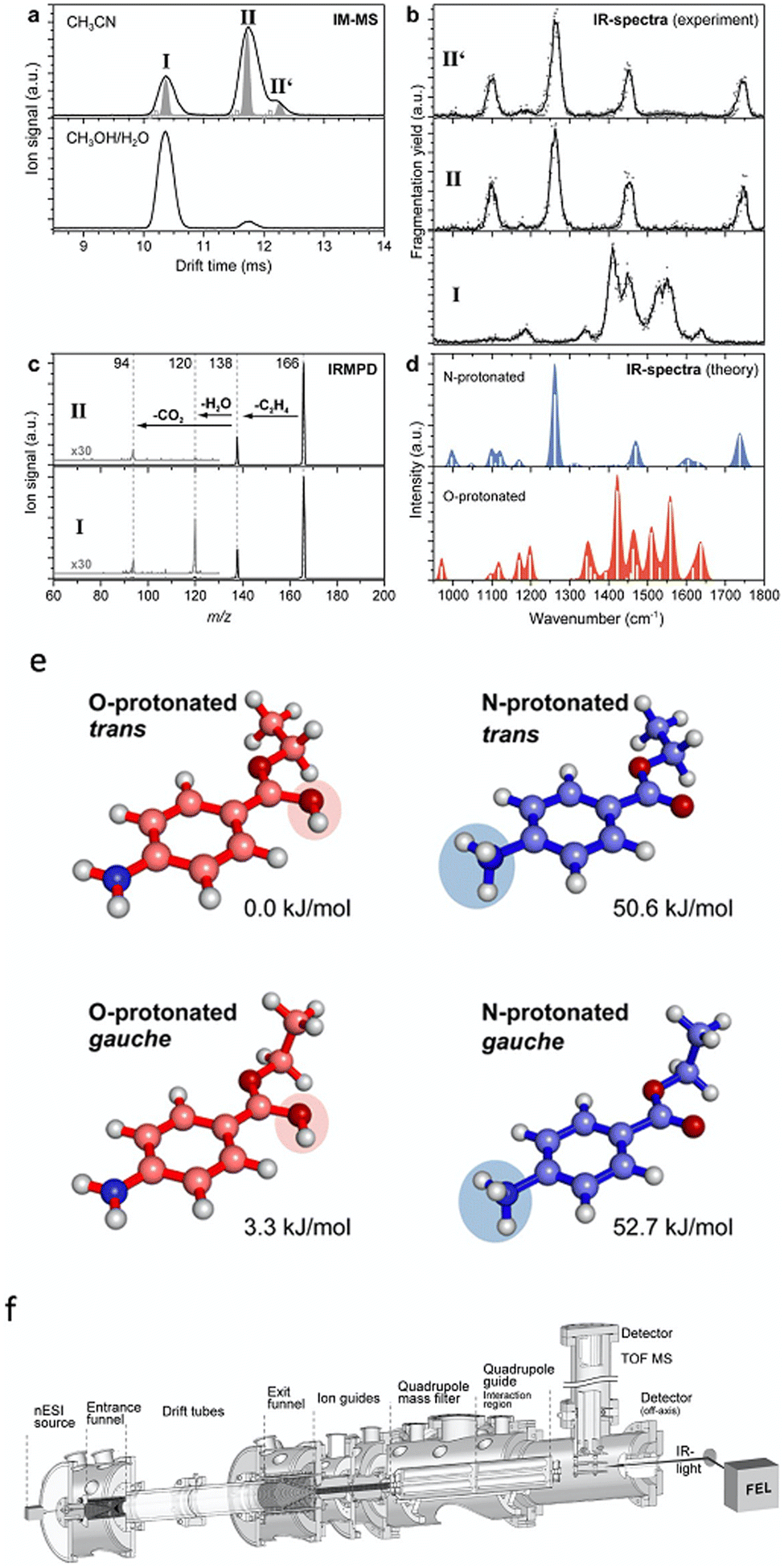 | ||
| Fig. 10 (a) Drift IMS for benzocaineH+ generated using acetonitrile or a mixture of methanol and water as the ESI solvent. (b) IRMPD spectra of the ion-mobility selected protomers and isomers of benzocaine-H+. (c) IRMPD mass spectra. (d) Calculated IR spectra for different benzocaineH+ protomers. Structures and relative ground state energies for different protomers and isomers of protonated benzocaine (e). Schematic of the experimental setup (f). Adapted from S. Warnke, J. Seo, J. Boschmans, F. Sobott, J. H. Scrivens, C. Bleiholder, M. T. Bowers, S. Gewinner, W. Schöllkopf and K. Pagel, J. Am. Chem. Soc., 2015, 137, 4236–4242. Original article is in the public domain.100 | ||
Ion mobility combined with room temperature IR PD has been used to distinguish isomers of sacharides,174 amino acids,175 amino acid clusters,176 small molecules,177 metal-oxide clusters,178 and β-sheet formation.179 Combined ion mobility and cryogenic IR PD studies have distinguished isomers of peptides180–182 and glycans.183–188 Gas-phase infrared spectroscopy of glycan isomers was also recently reviewed.189 In the case of many systems mentioned here, especially some glycan isomers, the difference between isomers is very slight and these isomers cannot always be distinguished using one technique alone (i.e. just ion mobility or just IR PD). It is only when ion mobility is combined with IR PD that these isomers can be clearly distinguished.
Another ion-mobility separation method is field-asymmetric ion mobility mass spectrometry (FAIMS).96,98,190 While traditional drift IMS separates ions by arrival time dictated by their cross-sectional mobility in a low-strength electric field, FAIMS filtered ions by mobility in both low and high-field strengths. The principles of FAIMS are described in detail elsewhere.191,192 Briefly, ions are carried by an inert gas between two electrodes across which a dispersion voltage (DV) is applied, which is an asymmetric voltage that oscillates between high and low voltage of opposite polarity. Ions are separated based on their displacement in the high field and low field regimes and different isomers will have different trajectories between the FAIMS electrodes. The trajectory of an isomer can be corrected by applying a compensation voltage (CV) so that only the selected isomer passes between FAIMS electrodes to be detected. The FAIMS voltages and their effect on the trajectory of an ion are shown schematically in Fig. 11D.
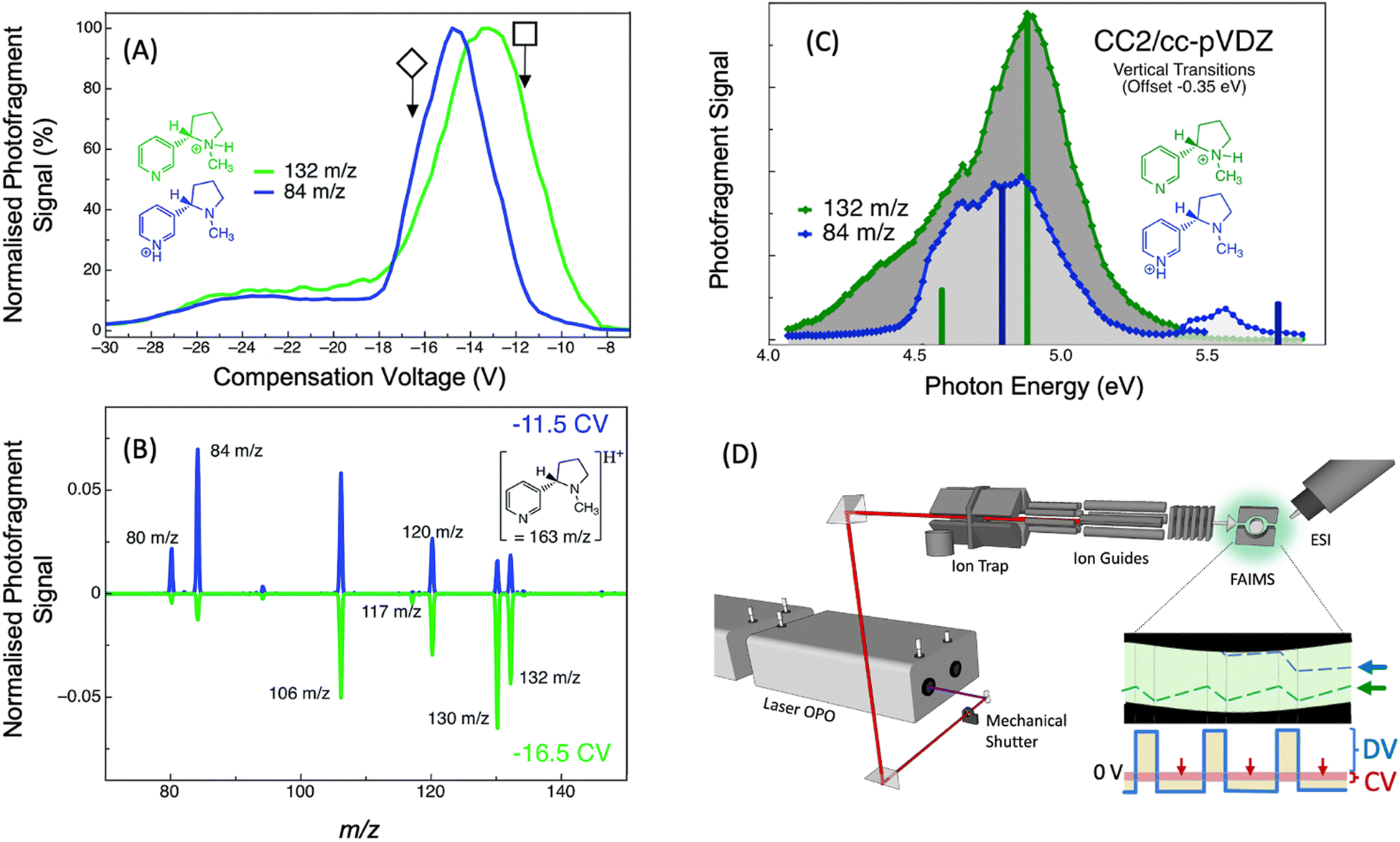 | ||
| Fig. 11 (A) FAIMS ionogram for nicotineH+ (m/z 163) following two PD products of nicotineH+ (m/z 132 and m/z 84), which are indicative of the two nicotine-H+ protomers. (B) PD mass spectra (λ = 266 nm) of nicotineH+ with FAIMS compensation voltage set to −11.5 V (blue line) or −16.5 V (green line). (C) Photodissociation action spectra of nicotineH+ following the signal of the m/z 84 photoproduct ion (blue trace) and the m/z 132 photoproduct ion (green trace). Vertical bars show vertical transition energies calculated using CC2 for the pyridine protonated protomer (blue bars) and the pyrolidine protonated protomer (green bars). Vertical transition energies are offset by −0.35 eV for comparison to experimental spectra. (D) Schematic illustration to provide a survey of the experimental arrangement at University of Wollongong. Ions are generated using ESI, the isomer can then be separated and selected using FAIMS, guided into the linear ion trap (LTQ) where they are m/z selected and stored before being irradiated by a laser pulse (red line) and scanned out of the ion trap to produce a mass spectrum. A schematic showing the DV (solid blue line) and CV (red shaded area) used in FAIMS and the trajectory that two different isomers might take (blue and green dashed lines) between the FAIMS electrodes. Reproduced from ref. 98 with permission from the Royal Society of Chemistry.98 | ||
Drift IMS and FAIMS each have advantages and disadvantages. While FAIMS can provide separation different from that of drift IMS,193 the FAIMS separation process is known to thermally activate ions194 and possibly cause structural rearrangements—this would be of particular concern for native biomolecular ion studies but less of a concern for small molecule investigations targeted here. Drift IMS provides collisional cross-section (CCS) values for separated species. These CCS values provide information on the ion shape and can be compared with other reference CCS values and calculated values. Therefore, even without PD spectroscopy, the CCS values can provide information on structural assignments of different isomers separated using IMS. A method to measure CCS values using FAIMS results is emerging,195 which is promising but not as established as CCS measurements based on drift IMS.
It is sometimes the case that protomer ions are not possible to disentangle without separation. In particular, broad featureless spectra can be difficult to deconvolve when there are multiple species with similar spectra. For example, in our study of nicotineH+, a combination of FAIMS and UV PD action spectroscopy (see Fig. 11D) was able to distinguish the overlapping spectra of the two nicotineH+ protomers.98 The UV PD action spectra of nicotineH+ (without FAIMS separation) are shown in Fig. 11C following the formation of the m/z 84 photoproduct (blue trace) and the m/z 132 photoproduct (green trace). The spectra obtained by following these two photoproducts are sufficiently different, with the green trace exhibiting a shoulder at a lower energy (<4.5 eV) and the blue trace exhibiting a high energy feature at 5.5 eV.
By comparing these experimental action spectra to vertical transition energies and relative intensities calculated with the CC2 method (vertical bars in Fig. 11C), it is found that there is good agreement between the calculated electronic transitions of the pyrrolidine protonated nicotineH+ and the spectrum obtained by following formation of the m/z 132 photoproduct and there is good agreement between the calculated electronic transitions of the pyridine protonated nicotineH+ and the spectrum obtained by following formation of the m/z 84 photoproduct. This is consistent with previous studies of the dissociation of nicotineH+ assigned to the m/z 132 product ion as forms from the pyrrolidine protomer of nicotineH+, while the m/z 84 product ion forms form the pyridine protomer of nicotineH+.196 Based on CC2 calculations, it was determined that the shoulder at <4.5 eV in the spectrum of the pyrrolidine protomer arises from an nπ* state, which involves the n-orbital from the pyridine-ring nitrogen's lone pair—this state is not present for the pyridine protomer because the pyridine lone pair is involved in the bonding to H+. Therefore, when compared to vertical electronic transitions, the UV PD action spectra provide direct spectroscopic evidence for the presence of two protomers of nicotineH+ and direct spectroscopic assignment of these two protomers.
A room temperature IRMPD spectrum of nicotineH+ has been reported,197 which concluded that just the pyridine-protonated protomer was present. This could signify that only one protomer was generated under those conditions of ESI, or perhaps both protomers were present and the spectrum of one was hidden under the spectrum of the other. Cryogenic infrared photodissociation spectra of H2 tagged nicotineH+ ions provide additional spectroscopic evidence for the presence of both nicotineH+ protomers.198
Another example of FAIMS separation of protomer ions is the case of quinazolineH+ where two protomers present from ESI in comparable yields.190 With FAIMS separation, the spectra of both protomers were separately recorded, with a vibronic structure (Fig. 12). The presence of this vibronic structure allows for compelling assignment of the isomer structures and this happens with Franck–Condon simulations and comparison to calculated 0–0 transition energies. In this case, the simulations nicely reproduce the experimental vibronic structure and the 0–0 energies for the S1 ← S0 transitions are measured as 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 cm−1 (calculated as 26650 cm−1 using MS-CASPT2) for 3-quinazolineH+ and 27600 cm−1 (calculated as 28424 cm−1 using MS-CASPT2) for 1-quinazolineH+. However, like the quinolineH+ and isoquinolineH+ spectra shown in Fig. 3, the 0–0 transition energies of quinazolineH+ protomers are close together (<1000 cm−1) and so require the use of an accurate method (in this case MS-CASPT2) to confidently make the assignment.
900 cm−1 (calculated as 26650 cm−1 using MS-CASPT2) for 3-quinazolineH+ and 27600 cm−1 (calculated as 28424 cm−1 using MS-CASPT2) for 1-quinazolineH+. However, like the quinolineH+ and isoquinolineH+ spectra shown in Fig. 3, the 0–0 transition energies of quinazolineH+ protomers are close together (<1000 cm−1) and so require the use of an accurate method (in this case MS-CASPT2) to confidently make the assignment.
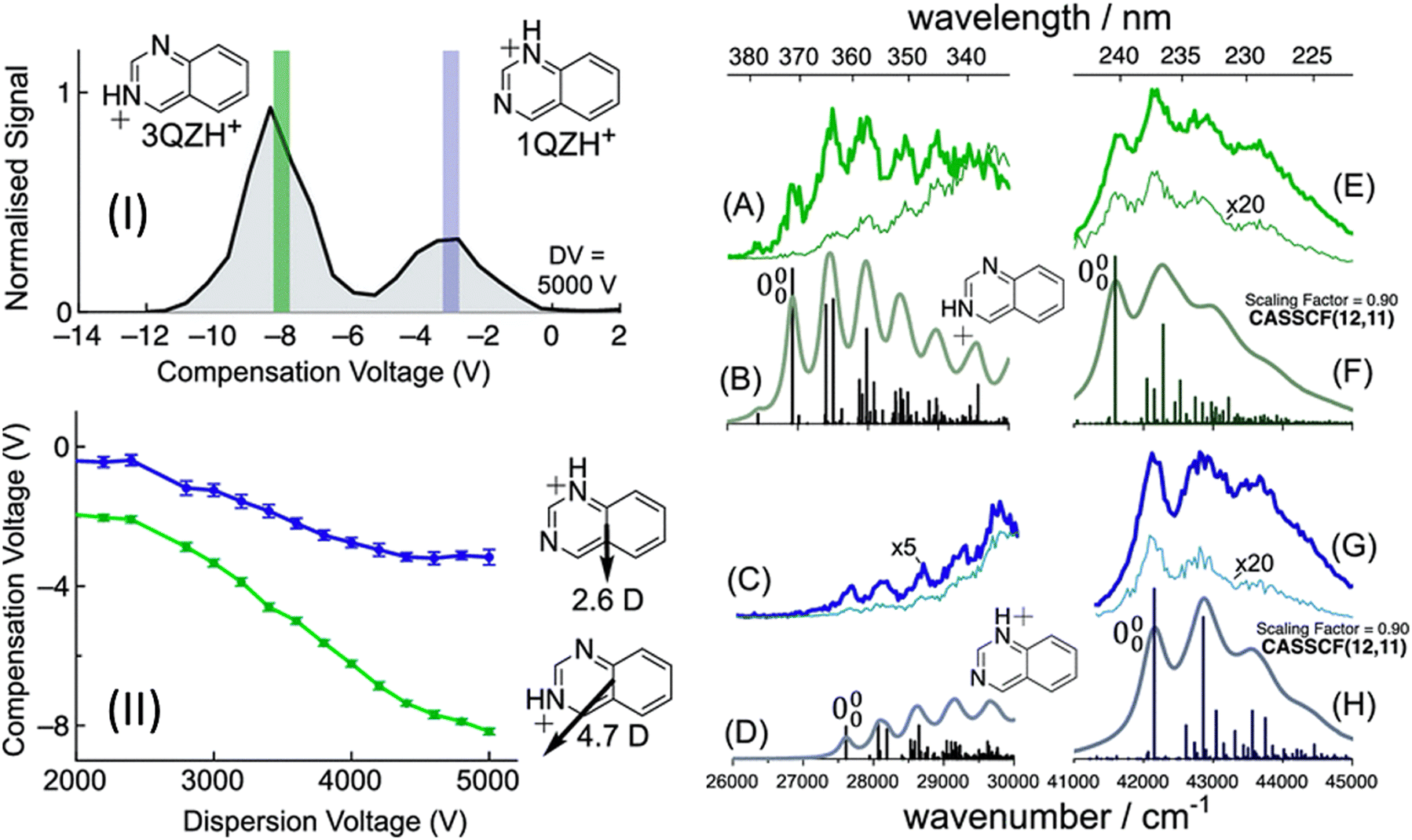 | ||
| Fig. 12 (I) FAIMS ionogram following quinazoline-H+ (m/z 131). Action spectra of quinazoline-H+ using compensation voltages of −8 V (A and E) or −3 V (C and G). Franck–Condon simulations for the S1 (La) state of 3-quinazoline-H+ (B) and 1-quinazoline-H+ (D). Franck–Condon simulations for the S5 (Bb) state of 3-quinazoline-H+ (F) and 1-quinazoline-H+ (H). S. J. Marlton, B. I. McKinnon, B. Ucur, J. P. Bezzina, S. J. Blanksby and A. J. Trevitt, J. Phys. Chem. Lett., 2020, 11, 4226–4231. Copyright 2020 American Chemical Society.190 | ||
Other examples of experiments that combine ion mobility with PD action spectroscopy are FAIMS separation with cryogenic UV/vis PD spectroscopy of peptides from Rizzo's group,180,199,200 FAIMS separation with room temperature UV/vis PD spectroscopy from the Hopkins group,96,201–204 drift mobility separation with cryogenic spectroscopy from Bieske's group,158,159 and drift mobility separation with room temperature photodissociation and photoisomerization from Bieske's group72,205–218 and Dugourd's group.219,220
Tandem IMS and photoactivation
Tandem IMS has been combined with photoactivation to observe photoisomerism. This is possible because photoisomerism can alter the ion CCS. The Bieske group72,205–218 and the Dugourd group219,220 have both deployed this strategy. The tandem ion mobility setup uses an IMS-IMS-QMF configuration with two drift ion mobility regions (IMS1 and IMS2). A schematic of this experimental setup is shown in Fig. 13, along with an ion mobility spectrum and photoisomerization action spectrum of protonated trans azobenzene reproduced from ref. 207. The trans isomer of protonated azobenzene is selected in the first ion mobility step (IMS1) so only this isomer is present. This is confirmed by the IMS2 arrival time distribution, which displays only one peak (black trace in Fig. 13a). If the mobility selected ions are irradiated by a laser between IMS1 and IMS2, photoisomerism can be induced as revealed by the signal from the cis isomer and the depletion of the trans (dotted line in Fig. 13b). The cis isomer peak is assigned by its arrival time from the IMS2 spectrum.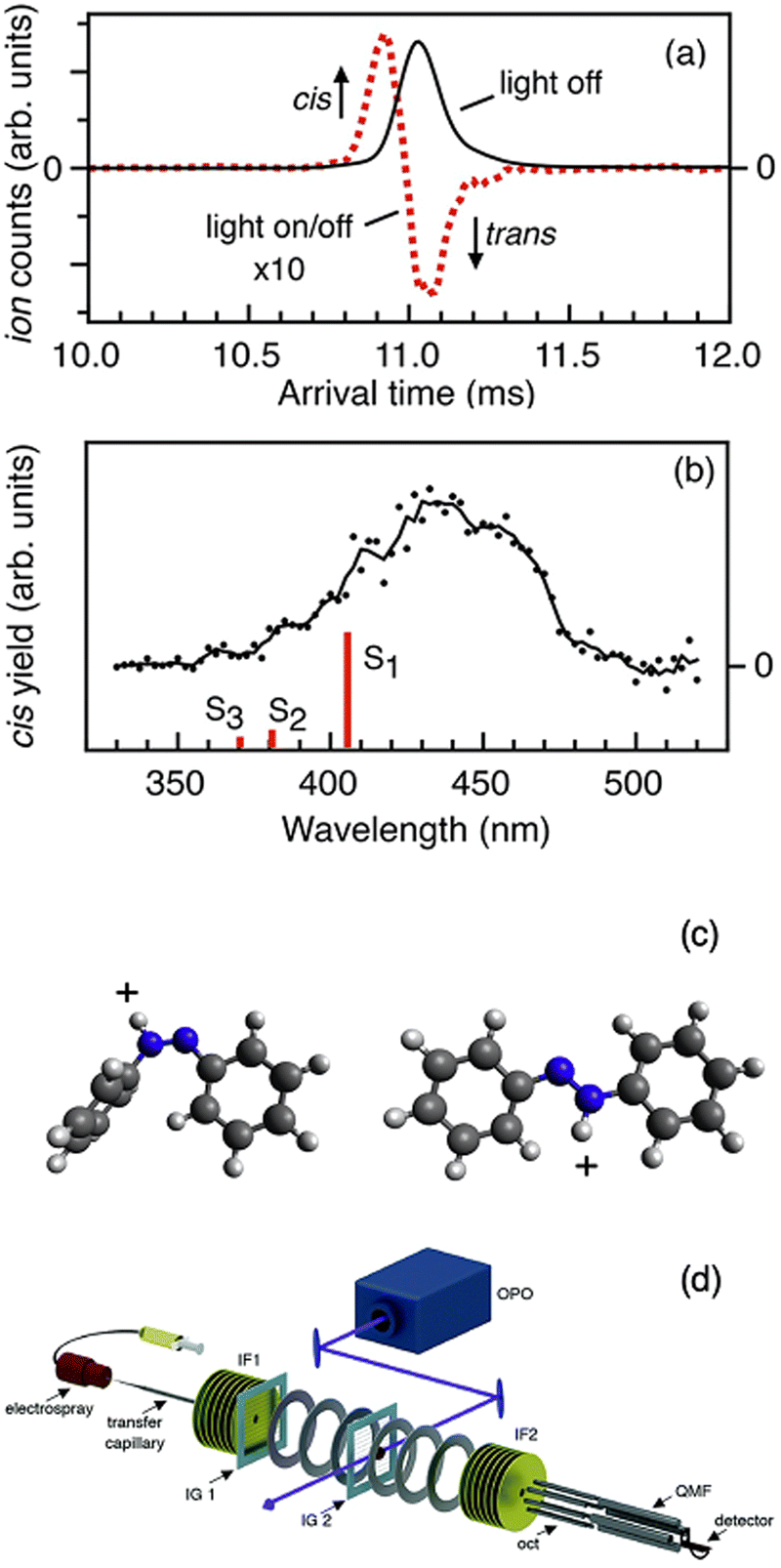 | ||
| Fig. 13 (a) Arrival time distributions measured by the IMS2 drift region for trans azobenzene (that was selected by its mobility in the IMS1 region). The traces correspond to light off (black trace) and 440 nm photoaction (red dashes). (b) Action spectrum following the photoisomerization to the cis isomer as a function of wavelength. (c) Structures of protonated cis azobenzene and trans azobenzene. (d) Schematic of the tandem ion-mobility experimental setup. (a), (b) and (d) Adapted with permission from M. S. Scholz, J. N. Bull, N. J. Coughlan, E. Carrascosa, B. D. Adamson and E. J. Bieske, J. Phys. Chem. A, 2017, 121, 6413–6419.207 | ||
Tandem ion mobility makes up a major proportion of the studies using combined ion-mobility and UV/vis action spectroscopy due to studies by the Bieske group72,205–218 as well as the Dugourd group.219,220 The example of protonated azobenzene highlights several key points about this technique. Ion mobility selection can allow for isomer specific investigation, tandem ion mobility can be used to study photoisomerism and the connection between conformational isomers.
Chirality
Chirality affects the biomolecule function including physiological and toxicological behavior.221 Conventional circular dichroism (CD) spectroscopy takes advantage of the fact that stereoisomers have different absorption values for left and right circularly polarised light. The development of gas phase PD techniques for distinguishing stereoisomers has emerged in recent years. The inherent advantages of coupling CD analysis with mass spectrometry include less sample quantity, lower sample concentration, and reduced purity concerns (the target can be m/z-isolated within the mass spectrometer) compared to conventional liquid-phase CD analysis.Recently, experimental gas phase PD arrangements have successfully distinguished between stereoisomers of small chiral ions with linearly polarised light.222,223 Experimental setups using circular polarised light to generate gas-phase CD action spectra have also been developed recently. Gas-phase CD action spectra have been reported for small ions in a cryogenic ion trap224,225 and large DNA polyanions at room temperature.226 Photoelectron circular dichroism of amino acid anions was also recently reported.227
A recent report, shown in Fig. 14, by Gabelica and co-workers described gas-phase chiral analysis where the signal is the normalised difference in electron photodetachment (ePD) yield arising from measurements alternating between left and right circularly polarised light. This is compared with the conventional solution phase CD spectrum (solid lines in Fig. 14). This approach demonstrates how gas phase laser and mass spectrometry techniques can be used to measure CD action spectra of selected ions.226 It is early days for this application but the incorporation of CD analysis coupled embedded in an ‘omics mass spectrometry is an alluring prospect.
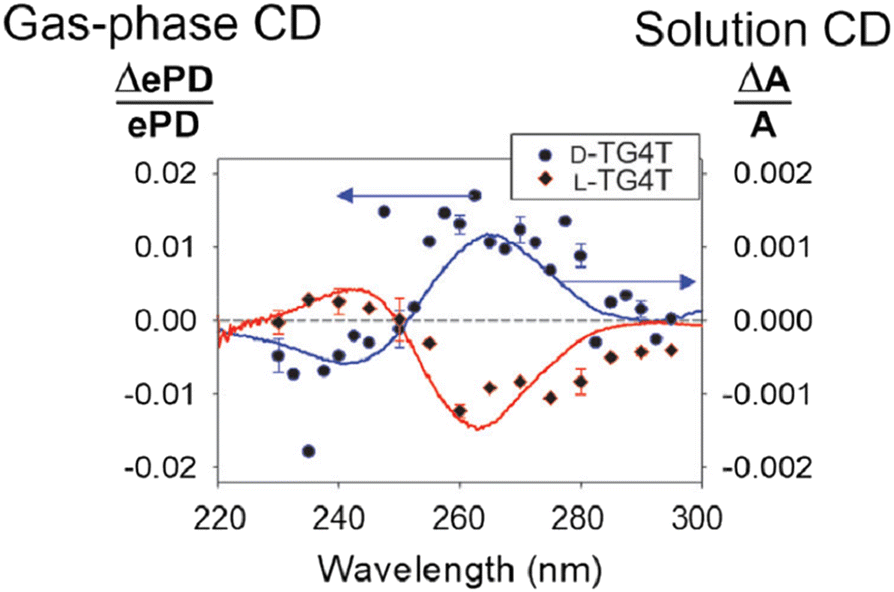 | ||
| Fig. 14 Gas-phase circular dichroism spectrum of m/z-selected right-handed G-quadruplex (blue data points) and left-handed chiral isomer (red data points) compared to conventional solution phase circular dichroism spectrum (solid lines). Adapted with permission from V. Gabelica, Accounts of Chemical Research, 2021, 54, 3691–3699.228 | ||
Final thoughts
Since the presence of isomers poses an outstanding challenge for many areas of mass spectrometry, it remains that more technical advances are required before isomer separation and disentanglement become routine. In this Feature Article, we focused on the techniques for distinguishing isomers that involve UV and visible photodissociation with mass spectrometry. These strategies are also applicable to the study of metal-cluster topological isomers like Au25(SR)18 (thiol-capped gold clusters),229 which can be liberated into the gas phase with ESI and separated with ion-mobility.230–232 Similar clusters have also been investigated with gas-phase techniques including photoelectron spectroscopy233 and photodissociation234 (but without isomer selection). Many of these experimental strategies are exciting prospects for further deployment and refinement. Incorporation of these techniques into routine mass spectrometry workflows remains an exciting avenue for exploitation. Furthermore, the application of mass spectrometry to areas including disease diagnostics, pathology and tissue imaging, and the numerous omics applications only further highlights the rich chemical information that will be available when isomer differentiation becomes commonplace. It was our intention to show here that photodissociation action spectroscopy strategies can provide definitive insights for a significant number of isomer classes. With suitable technical advancements, and reductions in size and cost of laser and optical hardware, it is easy to envisage a future where commercial mass spectrometers are equipped with tunable lasers affording visible, UV and VUV photoactivation. With these capabilities, PD action spectroscopy will be a vital tool to characterise the role of isomers in many areas including biological processes, physiology and the characterisation of diagnosis of disease.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Australian Research Council is gratefully acknowledged for the support for this research program (DP200100065, LE180100060). S. J. P. M. acknowledges support provided by the Australian Government Research Training Program Scholarship and support from UOW. We acknowledge the talents and dedicated efforts of all present and past students and researchers of the Laser Chemistry Laboratory at UOW. AJT is also grateful for the enduring and productive collaborations with Prof. SJ Blanksby and Dr BLJ Poad (QUT).References
- K. Uppal, D. I. Walker, K. Liu, S. Li, Y.-M. Go and D. P. Jones, Chem. Res. Toxicol., 2016, 29, 1956–1975 Search PubMed.
- S. J. Blanksby and T. W. Mitchell, Annu. Rev. Anal. Chem., 2010, 3, 433–465 Search PubMed.
- M. R. Paine, B. L. Poad, G. B. Eijkel, D. L. Marshall, S. J. Blanksby, R. M. Heeren and S. R. Ellis, Angew. Chem., Int. Ed., 2018, 57, 10530–10534 CrossRef CAS PubMed.
- Q. Wu, J.-Y. Wang, D.-Q. Han and Z.-P. Yao, TrAC, Trends Anal. Chem., 2020, 124, 115801 CrossRef CAS.
- M. Demireva and P. Armentrout, J. Phys. Chem. A, 2021, 125, 2849–2865 CrossRef CAS PubMed.
- A. K. Lam and R. A. O'Hair, Rapid Commun. Mass Spectrom., 2010, 24, 1779–1790 CrossRef CAS PubMed.
- J. L. Campbell, A. M.-C. Yang, L. R. Melo and W. S. Hopkins, J. Am. Soc. Mass Spectrom., 2016, 27, 1277–1284 CrossRef CAS PubMed.
- M. C. Thomas, T. W. Mitchell, D. G. Harman, J. M. Deeley, J. R. Nealon and S. J. Blanksby, Anal. Chem., 2008, 80, 303–311 CrossRef CAS PubMed.
- E. Matthews and C. E. Dessent, Phys. Chem. Chem. Phys., 2017, 19, 17434–17440 RSC.
- J. S. Brodbelt, Chem. Soc. Rev., 2014, 43, 2757–2783 RSC.
- R. C. Dunbar and E. W. Fu, J. Am. Chem. Soc., 1973, 95, 2716–2718 CrossRef CAS.
- B. Freiser and J. Beauchamp, J. Am. Chem. Soc., 1977, 99, 3214–3225 CrossRef CAS.
- R. C. Dunbar, Int. J. Mass Spectrom., 2015, 377, 159–171 CrossRef CAS.
- R. C. Dunbar, J. Am. Chem. Soc., 1971, 93, 4354–4358 CrossRef CAS.
- A. El-Aneed, A. Cohen and J. Banoub, Appl. Spectrosc. Rev., 2009, 44, 210–230 CrossRef CAS.
- L. Konermann, E. Ahadi, A. D. Rodriguez and S. Vahidi, Anal. Chem., 2013, 85, 2–9 CrossRef CAS PubMed.
- H. Awad, M. M. Khamis and A. El-Aneed, Appl. Spectrosc. Rev., 2015, 50, 158–175 CrossRef CAS.
- R. E. van Outersterp, J. Martens, G. Berden, V. Koppen, F. Cuyckens and J. Oomens, Analyst, 2020, 145, 6162–6170 RSC.
- M. G. Münst, M. Ončák, M. K. Beyer and C. van der Linde, J. Chem. Phys., 2021, 154, 084301 CrossRef PubMed.
- A. F. Cruz-Ortiz, R. A. Jara-Toro, M. Berdakin, E. Loire and G. A. Pino, Eur. Phys. J. D, 2021, 75, 1–11 CrossRef.
- A. P. Cismesia, G. R. Nicholls and N. C. Polfer, J. Mol. Spectrosc., 2017, 332, 79–85 CrossRef CAS PubMed.
- B. Schindler, G. Laloy-Borgna, L. Barnes, A.-R. Allouche, E. Bouju, V. Dugas, C. Demesmay and I. Compagnon, Anal. Chem., 2018, 90, 11741–11745 CrossRef CAS PubMed.
- R. Wu, L. Hamlow, C. He, Y.-W. Nei, G. Berden, J. Oomens and M. Rodgers, J. Am. Soc. Mass Spectrom., 2017, 28, 1638–1646 CrossRef CAS PubMed.
- J. Laskin and J. H. Futrell, Mass Spectrom. Rev., 2005, 24, 135–167 CrossRef CAS PubMed.
- J. S. Brodbelt and J. J. Wilson, Mass Spectrom. Rev., 2009, 28, 390–424 CrossRef CAS PubMed.
- S. Mitra, C. H. Duong, L. M. McCaslin, R. B. Gerber and M. A. Johnson, Phys. Chem. Chem. Phys., 2020, 22, 4501–4507 RSC.
- E. H. Perez, F. S. Menges, M. Cattaneo, J. M. Mayer and M. A. Johnson, J. Chem. Phys., 2020, 152, 234309 CrossRef CAS PubMed.
- K. Chatterjee and O. Dopfer, Astrophys. J., 2020, 898, 92 CrossRef CAS.
- E. Andris, R. Navrátil, J. Jašìk, T. Terencio, M. Srnec, M. Costas and J. Roithová, J. Am. Chem. Soc., 2017, 139, 2757–2765 CrossRef CAS PubMed.
- A. F. DeBlase, C. P. Harrilal, J. T. Lawler, N. L. Burke, S. A. McLuckey and T. S. Zwier, J. Am. Chem. Soc., 2017, 139, 5481–5493 CrossRef CAS PubMed.
- G. Z. Zhu, C. H. Qian and L. S. Wang, Angew. Chem., Int. Ed., 2019, 58, 7856–7860 CrossRef CAS PubMed.
- M. L. Weichman, J. A. DeVine, D. S. Levine, J. B. Kim and D. M. Neumark, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1698–1705 CrossRef CAS PubMed.
- J. N. Bull, C. S. Anstöter and J. R. Verlet, J. Phys. Chem. A, 2020, 124, 2140–2151 CrossRef CAS PubMed.
- J. L. Woodhouse, M. Assmann, M. A. Parkes, H. Grounds, S. J. Pacman, J. C. Anderson, G. A. Worth and H. H. Fielding, Phys. Chem. Chem. Phys., 2017, 19, 22711–22720 RSC.
- V. Dryza, N. Chalyavi, J. A. Sanelli and E. J. Bieske, J. Chem. Phys., 2012, 137, 204304 CrossRef PubMed.
- U. Jacovella, G. da Silva and E. J. Bieske, J. Phys. Chem. A, 2019, 123, 823–830 CrossRef CAS PubMed.
- U. Jacovella, M. S. Scholz and E. J. Bieske, J. Phys. Chem. Lett., 2020, 11, 8867–8872 CrossRef CAS PubMed.
- O. Krechkivska, Y. Liu, K. L. K. Lee, K. Nauta, S. H. Kable and T. W. Schmidt, J. Phys. Chem. Lett., 2013, 4, 3728–3732 CrossRef CAS.
- V. Gabelica and E. Marklund, Curr. Opin. Chem. Biol., 2018, 42, 51–59 CrossRef CAS PubMed.
- F. Lanucara, S. W. Holman, C. J. Gray and C. E. Eyers, Nat. Chem., 2014, 6, 281–294 CrossRef CAS PubMed.
- V. d'Atri, T. Causon, O. Hernandez-Alba, A. Mutabazi, J. L. Veuthey, S. Cianferani and D. Guillarme, J. Sep. Sci., 2018, 41, 20–67 CrossRef PubMed.
- R. C. Dunbar, Int. J. Mass Spectrom., 2000, 200, 571–589 CrossRef CAS.
- H. J. Zeng, N. Yang and M. A. Johnson, Faraday Discuss., 2019, 217, 8–33 RSC.
- F. Tureček, Mass Spectrom. Rev., 2021, 1–21 Search PubMed.
- T. L. Guasco and M. A. Johnson, Emerging Trends in Chemical Applications of Lasers, ACS Publications, 2021, pp. 277–306 Search PubMed.
- S. Soorkia, C. Jouvet and G. Grégoire, Chem. Rev., 2019, 120, 3296–3327 CrossRef PubMed.
- F. Tureček, J. Phys. Chem. B, 2021, 125, 7090–7100 CrossRef PubMed.
- H. Zettergren, A. Domaracka, T. Schlathölter, P. Bolognesi, S. Díaz-Tendero, M. Łabuda, S. Tosic, S. Maclot, P. Johnsson and A. Steber, Eur. Phys. J. D, 2021, 75, 1–53 CrossRef.
- R. Antoine and P. Dugourd, Phys. Chem. Chem. Phys., 2011, 13, 16494–16509 RSC.
- N. G. Hendricks and R. R. Julian, Analyst, 2016, 141, 4534–4540 RSC.
- J. P. Reilly, Mass Spectrom. Rev., 2009, 28, 425–447 CrossRef CAS PubMed.
- T. R. Rizzo, J. A. Stearns and O. V. Boyarkin, Int. Rev. Phys. Chem., 2009, 28, 481–515 Search PubMed.
- O. V. Boyarkin and V. Kopysov, Rev. Sci. Instrum., 2014, 85, 033105 CrossRef PubMed.
- C. S. Hansen, S. J. Blanksby and A. J. Trevitt, Phys. Chem. Chem. Phys., 2015, 17, 25882–25890 RSC.
- B. I. McKinnon, S. J. Marlton, B. Ucur, E. J. Bieske, B. L. Poad, S. J. Blanksby and A. J. Trevitt, J. Phys. Chem. Lett., 2021, 12, 11939–11944 CrossRef CAS PubMed.
- A. Dang, J. A. Korn, J. Gladden, B. Mozzone and F. Tureček, J. Am. Soc. Mass Spectrom., 2019, 30, 1558–1564 CrossRef CAS PubMed.
- C. S. Hansen, B. B. Kirk, S. J. Blanksby, R. A. O’Hair and A. J. Trevitt, J. Am. Soc. Mass Spectrom., 2013, 24, 932–940 CrossRef CAS PubMed.
- E. Matthews, A. Sen, N. Yoshikawa, E. Bergström and C. E. Dessent, Phys. Chem. Chem. Phys., 2016, 18, 15143–15152 RSC.
- Q. Bian, M. W. Forbes, F. O. Talbot and R. A. Jockusch, Phys. Chem. Chem. Phys., 2010, 12, 2590–2598 RSC.
- T. Ly and R. R. Julian, J. Am. Chem. Soc., 2008, 130, 351–358 CrossRef CAS PubMed.
- M. H. Stockett, J. Houmøller, K. Støchkel, A. Svendsen and S. Brøndsted Nielsen, Rev. Sci. Instrum., 2016, 87, 053103 CrossRef PubMed.
- N. J. Coughlan, P. J. Carr, S. C. Walker, C. Zhou, M. Guna, J. L. Campbell and W. S. Hopkins, J. Am. Soc. Mass Spectrom., 2020, 23, 20607–20614 Search PubMed.
- A. Günther, P. Nieto, D. Müller, A. Sheldrick, D. Gerlich and O. Dopfer, J. Mol. Spectrosc., 2017, 332, 8–15 CrossRef.
- I. Alata, J. Bert, M. Broquier, C. Dedonder, G. Féraud, G. Grégoire, S. Soorkia, E. Marceca and C. Jouvet, J. Phys. Chem. A, 2013, 117, 4420–4427 CrossRef CAS PubMed.
- S.-i Ishiuchi, H. Wako, D. Kato and M. Fujii, J. Mol. Spectrosc., 2017, 332, 45–51 CrossRef CAS.
- H. Kang, G. Féraud, C. Dedonder-Lardeux and C. Jouvet, J. Phys. Chem. Lett., 2014, 5, 2760–2764 CrossRef CAS PubMed.
- X.-B. Wang and L.-S. Wang, Rev. Sci. Instrum., 2008, 79, 073108 CrossRef PubMed.
- S. Xu, S. Gozem, A. I. Krylov, C. R. Christopher and J. M. Weber, Phys. Chem. Chem. Phys., 2015, 17, 31938–31946 RSC.
- J. Jasik, R. Navrátil, I. Němec and J. Roithová, J. Phys. Chem. A, 2015, 119, 12648–12655 CrossRef CAS PubMed.
- J. G. Redwine, Z. A. Davis, N. L. Burke, R. A. Oglesbee, S. A. McLuckey and T. S. Zwier, Int. J. Mass Spectrom., 2013, 348, 9–14 CrossRef CAS.
- B. Adamson, N. Coughlan, R. Continetti and E. Bieske, Phys. Chem. Chem. Phys., 2013, 15, 9540–9548 RSC.
- B. Adamson, N. Coughlan, P. Markworth, R. Continetti and E. Bieske, Rev. Sci. Instrum., 2014, 85, 123109 CrossRef CAS PubMed.
- B. Bellina, J. M. Brown, J. Ujma, P. Murray, K. Giles, M. Morris, I. Compagnon and P. E. Barran, Analyst, 2014, 139, 6348–6351 RSC.
- F. Misaizu, N. Hori, H. Tanaka, K. Komatsu, A. Furuya and K. Ohno, Eur. Phys. J. D, 2009, 52, 59–62 CrossRef CAS.
- J. U. Andersen, P. Hvelplund, S. B. Nielsen, S. Tomita, H. Wahlgreen, S. P. Møller, U. V. Pedersen, J. S. Forster and T. J. Jørgensen, Rev. Sci. Instrum., 2002, 73, 1284–1287 CrossRef CAS.
- R. D. Thomas, H. T. Schmidt, G. Andler, M. Björkhage, M. Blom, L. Brännholm, E. Bäckström, H. Danared, S. Das and N. Haag, Rev. Sci. Instrum., 2011, 82, 065112 CrossRef CAS PubMed.
- A. D. Depland, G. Renois-Predelus, B. Schindler and I. Compagnon, Int. J. Mass Spectrom., 2018, 434, 65–69 CrossRef.
- L. Jaravel, B. Schindler, J. Randon, I. Compagnon, C. Demesmay and V. Dugas, J. Chromatogr. A, 2020, 1617, 460782 CrossRef CAS PubMed.
- M. Broquier, S. Soorkia, C. Dedonder-Lardeux, C. Jouvet, P. Theulé and G. Grégoire, J. Phys. Chem. A, 2016, 120, 3797–3809 CrossRef CAS PubMed.
- R. L. Garcia, N. Nieuwjaer, C. Desfrançois, F. Lecomte, S. Leite, B. Manil, M. Broquier and G. Grégoire, Phys. Chem. Chem. Phys., 2017, 19, 8258–8268 RSC.
- S. Soorkia, M. Broquier and G. Grégoire, Phys. Chem. Chem. Phys., 2016, 18, 23785–23794 RSC.
- J. P. Bezzina, MPhil thesis, School of Chemistry, University of Wollongong, 2018.
- J. A. Noble, E. Marceca, C. Dedonder, W. Phasayavan, G. Féraud, B. Inceesungvorn and C. Jouvet, Phys. Chem. Chem. Phys., 2020, 22, 27280–27289 RSC.
- J. A. Noble, E. Marceca, C. Dedonder and C. Jouvet, Phys. Chem. Chem. Phys., 2020, 22, 27290–27299 RSC.
- G. A. Pino, G. Feraud, M. Broquier, G. Grégoire, S. Soorkia, C. Dedonder and C. Jouvet, Phys. Chem. Chem. Phys., 2016, 18, 20126–20134 RSC.
- C. S. Hansen, PhD thesis, School of Chemistry, University of Wollongong, 2016.
- S. J. Marlton, B. I. McKinnon, P. Greißel, O. J. Shiels, B. Ucur and A. J. Trevitt, J. Chem. Phys., 2021, 155, 184302 CrossRef CAS PubMed.
- L. G. Dodson, W. Zagorec-Marks, S. Xu, J. E. Smith and J. M. Weber, Phys. Chem. Chem. Phys., 2018, 20, 28535–28543 RSC.
- M. Wanko, J. Houmøller, K. Støchkel, M.-B. S. Kirketerp, M. Å. Petersen, M. B. Nielsen, S. B. Nielsen and A. Rubio, Phys. Chem. Chem. Phys., 2012, 14, 12905–12911 RSC.
- J. Houmøller, M. Wanko, A. Rubio and S. B. Nielsen, J. Phys. Chem. A, 2015, 119, 11498–11503 CrossRef PubMed.
- K. Uleanya and C. E. H. Dessent, Phys. Chem. Chem. Phys., 2021, 23, 1021–1030 RSC.
- G. Féraud, L. Domenianni, E. Marceca, C. Dedonder-Lardeux and C. Jouvet, J. Phys. Chem. A, 2017, 121, 2580–2587 CrossRef PubMed.
- P.-F. o Loos, N. Galland and D. Jacquemin, J. Phys. Chem. Lett., 2018, 9, 4646–4651 CrossRef CAS PubMed.
- P.-F. Loos and D. Jacquemin, J. Chem. Theory Comput., 2019, 15, 2481–2491 CrossRef CAS PubMed.
- P.-F. Loos, A. Scemama and D. Jacquemin, J. Phys. Chem. Lett., 2020, 11, 2374–2383 CrossRef CAS PubMed.
- N. J. Coughlan, P. J. Carr, S. C. Walker, C. Zhou, M. Guna, J. L. Campbell and W. S. Hopkins, J. Am. Soc. Mass Spectrom., 2020, 31, 405–410 CrossRef CAS PubMed.
- H. Xia and A. B. Attygalle, J. Am. Soc. Mass Spectrom., 2017, 28, 2580–2587 CrossRef CAS PubMed.
- S. J. Marlton, B. I. McKinnon, B. Ucur, A. T. Maccarone, W. A. Donald, S. J. Blanksby and A. J. Trevitt, Faraday Discuss., 2019, 217, 453–475 RSC.
- J. Chandran, Z. Zheng, V. I. Thomas, C. Rajalakshmi and A. B. Attygalle, Analyst, 2020, 145, 5333–5344 RSC.
- S. Warnke, J. Seo, J. Boschmans, F. Sobott, J. H. Scrivens, C. Bleiholder, M. T. Bowers, S. Gewinner, W. Schöllkopf and K. Pagel, J. Am. Chem. Soc., 2015, 137, 4236–4242 CrossRef CAS PubMed.
- J. Schmidt, M. M. Meyer, I. Spector and S. R. Kass, J. Phys. Chem. A, 2011, 115, 7625–7632 CrossRef CAS PubMed.
- Z. Tian and S. R. Kass, Angew. Chem., 2009, 121, 1347–1349 CrossRef.
- E. Matthews, R. Cercola and C. Dessent, Molecules, 2018, 23, 2036 CrossRef PubMed.
- E. Matthews and C. E. Dessent, J. Phys. Chem. A, 2016, 120, 9209–9216 CrossRef CAS PubMed.
- M. Pitzer, C. Ozga, C. Küstner-Wetekam, P. Reiß, A. Knie, A. Ehresmann, T. Jahnke, A. Giuliani and L. Nahon, J. Phys. Chem. A, 2019, 123, 3551–3557 CrossRef CAS PubMed.
- S. Ø. Pedersen, C. S. Byskov, F. Turecek and S. B. Nielsen, J. Phys. Chem. A, 2014, 118, 4256–4265 CrossRef CAS PubMed.
- J. A. Berenbeim, N. G. Wong, M. C. Cockett, G. Berden, J. Oomens, A. M. Rijs and C. E. Dessent, J. Phys. Chem. A, 2020, 124, 2919–2930 CrossRef CAS PubMed.
- N. G. Wong, J. A. Berenbeim, M. Hawkridge, E. Matthews and C. E. Dessent, Phys. Chem. Chem. Phys., 2019, 21, 14311–14321 RSC.
- J. A. Berenbeim, N. G. Wong, M. C. Cockett, G. Berden, J. Oomens, A. M. Rijs and C. E. Dessent, Phys. Chem. Chem. Phys., 2020, 22, 19522–19531 RSC.
- S. R. Huang, A. Dang and F. E. Tureček, J. Am. Soc. Mass Spectrom., 2020, 31, 1271–1281 CrossRef CAS PubMed.
- M. Lesslie, J. T. Lawler, A. Dang, J. A. Korn, D. Bím, V. Steinmetz, P. Maître, F. Tureček and V. Ryzhov, Chem. Phys. Chem., 2017, 18, 1293–1301 CrossRef CAS PubMed.
- A. Dang, H. T. Nguyen, H. Ruiz, E. Piacentino, V. Ryzhov and F. E. Tureček, J. Phys. Chem. B, 2018, 122, 86–97 CrossRef CAS PubMed.
- A. Dang, Y. Liu and F. E. Tureček, J. Phys. Chem. A, 2019, 123, 3272–3284 CrossRef CAS PubMed.
- D. Müller and O. Dopfer, Phys. Chem. Chem. Phys., 2020, 22, 18328–18339 RSC.
- D. Müller and O. Dopfer, J. Photochem. Photobiol., 2020, 3, 100009 CrossRef.
- D. Müller, P. Nieto, M. Miyazaki and O. Dopfer, Faraday Discuss., 2019, 217, 256–275 RSC.
- M. Broquier, S. Soorkia, G. A. Pino, C. Dedonder-Lardeux, C. Jouvet and G. Gregoire, J. Phys. Chem. A, 2017, 121, 6429–6439 CrossRef CAS PubMed.
- M. Berdakin, G. Féraud, C. Dedonder-Lardeux, C. Jouvet and G. A. Pino, Phys. Chem. Chem. Phys., 2014, 16, 10643–10650 RSC.
- J. A. Noble, C. Dedonder-Lardeux, J. Mascetti and C. Jouvet, Chem. – Asian J., 2017, 12, 1523–1531 CrossRef CAS PubMed.
- J. A. Noble, M. Broquier, G. Gregoire, S. Soorkia, G. Pino, E. Marceca, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2018, 20, 6134–6145 RSC.
- E. Andris, R. Navratil, J. Jasik, M. Puri, M. Costas, L. Que Jr and J. Roithová, J. Am. Chem. Soc., 2018, 140, 14391–14400 CrossRef CAS PubMed.
- E. Andris, R. Navrátil, J. Jašík, G. Sabenya, M. Costas, M. Srnec and J. Roithová, Angew. Chem., Int. Ed., 2017, 56, 14057–14060 CrossRef CAS PubMed.
- R. Navrátil, S. Wiedbrauk, J. Jašík, H. Dube and J. Roithová, Phys. Chem. Chem. Phys., 2018, 20, 6868–6876 RSC.
- W. Zagorec-Marks, M. M. Foreman, J. R. Verlet and J. M. Weber, J. Phys. Chem. Lett., 2019, 10, 7817–7822 CrossRef CAS PubMed.
- E. Saparbaev, R. Yamaletdinov and O. V. Boyarkin, Anal. Chem., 2021, 93, 12822–12926 CrossRef CAS PubMed.
- E. Saparbaev, V. Kopysov, V. Aladinskaia, V. Ferrieres, L. Legentil and O. V. Boyarkin, Anal. Chem., 2020, 92, 14624–14632 CrossRef CAS PubMed.
- P. Nieto, D. Müller, A. Sheldrick, A. Günther, M. Miyazaki and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 22148–22158 RSC.
- G. Féraud, N. Esteves-Lopez, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2015, 17, 25755–25760 RSC.
- A. Patzer, M. Schütz, C. Jouvet and O. Dopfer, J. Phys. Chem. A, 2013, 117, 9785–9793 CrossRef CAS PubMed.
- I. Alata, M. Broquier, C. Dedonder, C. Jouvet and E. Marceca, Chem. Phys., 2012, 393, 25–31 CrossRef CAS.
- S. Soorkia, M. Broquier and G. Grégoire, J. Phys. Chem. Lett., 2014, 5, 4349–4355 CrossRef CAS PubMed.
- J. Dezalay, M. Broquier, S. Soorkia and G. Grégoire, Eur. Phys. J. D, 2021, 75, 1–11 CrossRef.
- J. M. Papanikolas, V. Vorsa, M. E. Nadal, P. J. Campagnola, J. R. Gord and W. Lineberger, J. Chem. Phys., 1992, 97, 7002–7005 CrossRef CAS.
- V. Vorsa, S. Nandi, P. J. Campagnola, M. Larsson and W. Lineberger, J. Chem. Phys., 1997, 106, 1402–1410 CrossRef CAS.
- A. Sanov, T. Sanford, S. Nandi and W. C. Lineberger, J. Chem. Phys., 1999, 111, 664–675 CrossRef CAS.
- M. Hervé, A. Boyer, R. Brédy, I. Compagnon, A.-R. Allouche and F. Lépine, Commun. Chem., 2021, 4, 1–7 CrossRef.
- A. Svendsen, H. V. Kiefer, H. B. Pedersen, A. V. Bochenkova and L. H. Andersen, J. Am. Chem. Soc., 2017, 139, 8766–8771 CrossRef CAS PubMed.
- H. V. Kiefer, E. Gruber, J. Langeland, P. A. Kusochek, A. V. Bochenkova and L. H. Andersen, Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
- H. Kang, C. Jouvet, C. Dedonder-Lardeux, S. Martrenchard, C. Charriere, G. Gregoire, C. Desfrancois, J. Schermann, M. Barat and J. Fayeton, J. Chem. Phys., 2005, 122, 084307 CrossRef CAS PubMed.
- H. Kang, C. Dedonder-Lardeux, C. Jouvet, G. Gregoire, C. Desfrancois, J.-P. Schermann, M. Barat and A. Fayeton, J. Phys. Chem. A, 2005, 109, 2417–2420 CrossRef CAS PubMed.
- H. Kang, C. Jouvet, C. Dedonder-Lardeux, S. Martrenchard, G. Gregoire, C. Desfrancois, J.-P. Schermann, M. Barat and J. Fayeton, Phys. Chem. Chem. Phys., 2005, 7, 394–398 RSC.
- J. Dezalay, M. Broquier, S. Soorkia, K. Hirata, S.-I. Ishiuchi, M. Fujii and G. Grégoire, Phys. Chem. Chem. Phys., 2020, 22, 11498–11507 RSC.
- F. Liedy, F. Bäppler, E. Waldt, Y. Nosenko, D. Imanbaew, A. Bhunia, M. Yadav, R. Diller, M. M. Kappes and P. W. Roesky, ChemPhysChem, 2018, 19, 3050–3060 CrossRef CAS PubMed.
- S. Kruppa, F. Bäppler, W. Klopper, S. Walg, W. Thiel, R. Diller and C. Riehn, Phys. Chem. Chem. Phys., 2017, 19, 22785–22800 RSC.
- S. V. Kruppa, F. Bäppler, C. Holzer, W. Klopper, R. Diller and C. Riehn, J. Phys. Chem. Lett., 2018, 9, 804–810 CrossRef CAS PubMed.
- L. MacAleese, S. Hermelin, K. E. Hage, P. Chouzenoux, A. Kulesza, R. Antoine, L. Bonacina, M. Meuwly, J.-P. Wolf and P. Dugourd, J. Am. Chem. Soc., 2016, 138, 4401–4407 CrossRef CAS PubMed.
- M. Bouakil, F. Chirot, M. Girod, P. Dugourd and L. MacAleese, Struct. Dyn., 2020, 7, 024302 CrossRef CAS PubMed.
- M. Bouakil, A. Kulesza, S. Daly, L. MacAleese, R. Antoine and P. Dugourd, J. Am. Soc. Mass Spectrom., 2017, 28, 2181–2188 CrossRef CAS PubMed.
- V. Scutelnic, A. Prlj, A. Zabuga, C. M. Corminboeuf and T. R. Rizzo, J. Phys. Chem. Lett., 2018, 9, 3217–3223 CrossRef CAS PubMed.
- A. V. Zabuga, M. Z. Kamrath, O. V. Boyarkin and T. R. Rizzo, J. Chem. Phys., 2014, 141, 10B609_601 CrossRef PubMed.
- N. Yang, S. C. Edington, T. H. Choi, E. V. Henderson, J. P. Heindel, S. S. Xantheas, K. D. Jordan and M. A. Johnson, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 26047–26052 CrossRef CAS PubMed.
- S.-i Ishiuchi, H. Wako, S. S. Xantheas and M. Fujii, Faraday Discuss., 2019, 217, 396–413 RSC.
- J. C. Dean, N. L. Burke, J. R. Hopkins, J. G. Redwine, P. Ramachandran, S. A. McLuckey and T. S. Zwier, J. Phys. Chem. A, 2015, 119, 1917–1932 CrossRef CAS PubMed.
- N. S. Nagornova, T. R. Rizzo and O. V. Boyarkin, Angew. Chem., 2013, 125, 6118–6121 CrossRef.
- J. A. Stearns, C. Seaiby, O. V. Boyarkin and T. R. Rizzo, Phys. Chem. Chem. Phys., 2009, 11, 125–132 RSC.
- O. V. Boyarkin, Int. Rev. Phys. Chem., 2018, 37, 559–606 Search PubMed.
- K. Koyasu, T. Ohtaki, N. Hori and F. Misaizu, Chem. Phys. Lett., 2012, 523, 54–59 CrossRef CAS.
- J. T. Buntine, M. I. Cotter, U. Jacovella, C. Liu, P. Watkins, E. Carrascosa, J. N. Bull, L. Weston, G. Muller and M. S. Scholz, J. Chem. Phys., 2021, 155, 214302 CrossRef CAS PubMed.
- J. T. Buntine, E. Carrascosa, J. N. Bull, U. Jacovella, M. I. Cotter, P. Watkins, C. Liu, M. S. Scholz, B. D. Adamson, S. J. P. Marlton and E. J. Bieske, Rev. Sci. Instrum., 2022, 93, 043201 CrossRef CAS PubMed.
- R. Fromherz, G. Ganteför and A. A. Shvartsburg, Phys. Rev. Lett., 2002, 89, 083001 CrossRef PubMed.
- R. Moriyama, T. Ohtaki, J. Hosoya, K. Koyasu and F. Misaizu, Eur. Phys. J. D, 2013, 67, 1–5 CrossRef.
- S. M. Zucker, S. Lee, N. Webber, S. J. Valentine, J. P. Reilly and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2011, 22, 1477–1485 CrossRef CAS PubMed.
- M. Vonderach, O. T. Ehrler, K. Matheis, P. Weis and M. M. Kappes, J. Am. Chem. Soc., 2012, 134, 7830–7841 CrossRef CAS PubMed.
- M. Vonderach, O. T. Ehrler, P. Weis and M. M. Kappes, Anal. Chem., 2011, 83, 1108–1115 CrossRef CAS PubMed.
- M. Vonderach, M.-O. Winghart, L. MacAleese, F. Chirot, R. Antoine, P. Dugourd, P. Weis, O. Hampe and M. M. Kappes, Phys. Chem. Chem. Phys., 2014, 16, 3007–3013 RSC.
- S. Warnke, C. Baldauf, M. T. Bowers, K. Pagel and G. von Helden, J. Am. Chem. Soc., 2014, 136, 10308–10314 CrossRef CAS PubMed.
- S. Warnke, G. von Helden and K. Pagel, Proteomics, 2015, 15, 2804–2812 CrossRef CAS PubMed.
- A.-L. Simon, F. Chirot, C. M. Choi, C. Clavier, M. Barbaire, J. Maurelli, X. Dagany, L. MacAleese and P. Dugourd, Rev. Sci. Instrum., 2015, 86, 094101 CrossRef PubMed.
- A. Theisen, B. Yan, J. M. Brown, M. Morris, B. Bellina and P. E. Barran, Anal. Chem., 2016, 88, 9964–9971 CrossRef CAS PubMed.
- K. A. Morrison and B. H. Clowers, J. Am. Soc. Mass Spectrom., 2017, 28, 1236–1241 CrossRef CAS PubMed.
- V. Frankevich, P. Martinez-Lozano Sinues, K. Barylyuk and R. Zenobi, Anal. Chem., 2013, 85, 39–43 CrossRef CAS PubMed.
- V. E. Frankevich, K. V. Barylyuk, P. M.-L. Sinues and R. Zenobi, J. Anal. Chem., 2014, 69, 1215–1219 CrossRef CAS.
- K. Guo, K. Ni, X. Song, K. Li, B. Tang, Q. Yu, X. Qian and X. Wang, Anal. Chem., 2018, 90, 4514–4520 CrossRef CAS PubMed.
- O. Hernandez, S. Isenberg, V. Steinmetz, G. L. Glish and P. Maitre, J. Phys. Chem. A, 2015, 119, 6057–6064 CrossRef CAS PubMed.
- F. Berthias, B. Maatoug, G. L. Glish, F. Moussa and P. Maitre, J. Am. Soc. Mass Spectrom., 2018, 29, 752–760 CrossRef CAS PubMed.
- J. Seo, S. Warnke, K. Pagel, M. T. Bowers and G. von Helden, Nat. Chem., 2017, 9, 1263 CrossRef CAS PubMed.
- B. Schindler, A. D. Depland, G. Renois-Predelus, G. Karras, B. Concina, G. Celep, J. Maurelli, V. Loriot, E. Constant and R. Bredy, Int. J. Ion Mobil. Spectrom., 2017, 20, 119–124 CrossRef CAS.
- M. Marianski, J. Seo, E. Mucha, D. A. Thomas, S. Jung, R. Schlögl, G. Meijer, A. Trunschke and G. von Helden, J. Phys. Chem. C, 2018, 123, 7845–7853 CrossRef.
- J. Seo, W. Hoffmann, S. Warnke, X. Huang, S. Gewinner, W. Schöllkopf, M. T. Bowers, G. von Helden and K. Pagel, Nat. Chem., 2017, 9, 39–44 CrossRef CAS PubMed.
- L. Voronina and T. R. Rizzo, Phys. Chem. Chem. Phys., 2015, 17, 25828–25836 RSC.
- A. Masson, M. Z. Kamrath, M. A. Perez, M. S. Glover, U. Rothlisberger, D. E. Clemmer and T. R. Rizzo, J. Am. Soc. Mass. Spectrom., 2015, 26, 1444–1454 CrossRef CAS PubMed.
- L. Voronina, A. Masson, M. Kamrath, F. Schubert, D. Clemmer, C. Baldauf and T. Rizzo, J. Am. Chem. Soc., 2016, 138, 9224–9233 CrossRef CAS PubMed.
- C. Masellis, N. Khanal, M. Z. Kamrath, D. E. Clemmer and T. R. Rizzo, J. Am. Soc. Mass Spectrom., 2017, 28, 2217–2222 CrossRef CAS PubMed.
- A. Ben Faleh, S. Warnke and T. R. Rizzo, Anal. Chem., 2019, 91, 4876–4882 CrossRef CAS PubMed.
- S. Warnke, A. Ben Faleh, V. Scutelnic and T. R. Rizzo, J. Am. Soc. Mass Spectrom., 2019, 30, 2204–2211 CrossRef CAS PubMed.
- P. Bansal, V. Yatsyna, A. H. AbiKhodr, S. Warnke, A. Ben Faleh, N. Yalovenko, V. H. Wysocki and T. R. Rizzo, Anal. Chem., 2020, 92, 9079–9085 CrossRef CAS PubMed.
- N. Yalovenko, V. Yatsyna, P. Bansal, A. H. AbiKhodr and T. R. Rizzo, Analyst, 2020, 145, 6493–6499 RSC.
- D. Kim Greis, E. Mucha, M. Lettow, D. A. Thomas, C. Kirschbaum, S. Moon, P. Alonso Pardo-Vargas, G. von Helden, G. Meijer and K. Gilmore, ChemPhysChem, 2020, 21, 1905 CrossRef PubMed.
- C. J. Gray, I. Compagnon and S. L. Flitsch, Curr. Opin. Struct. Biol., 2020, 62, 121–131 CrossRef CAS PubMed.
- S. J. Marlton, B. I. McKinnon, B. Ucur, J. P. Bezzina, S. J. Blanksby and A. J. Trevitt, J. Phys. Chem. Lett., 2020, 11, 4226–4231 CrossRef CAS PubMed.
- I. Buryakov, E. Krylov, E. Nazarov and U. K. Rasulev, Int. J. Mass Spectrom. Ion Processes, 1993, 128, 143–148 CrossRef CAS.
- A. A. Shvartsburg, Differential ion mobility spectrometry: nonlinear ion transport and fundamentals of FAIMS, CRC Press, 2008 Search PubMed.
- J. D. Zhang, M. T. Donor, A. D. Rolland, M. G. Leeming, H. Wang, A. J. Trevitt, K. M. Kabir, J. S. Prell and W. A. Donald, Int. J. Mass Spectrom., 2020, 457, 116425 CrossRef CAS.
- C. Ieritano, J. Featherstone, A. Haack, M. Guna, J. L. Campbell and W. S. Hopkins, J. Am. Soc. Mass Spectrom., 2020, 31, 582–593 CrossRef CAS PubMed.
- C. Ieritano, A. Lee, J. Crouse, Z. Bowman, N. Mashmoushi, P. M. Crossley, B. P. Friebe, J. L. Campbell and W. S. Hopkins, Anal. Chem., 2021, 93, 8937–8944 CrossRef CAS PubMed.
- J. P. Williams, N. M. Nibbering, B. N. Green, V. J. Patel and J. H. Scrivens, J. Mass Spectrom., 2006, 41, 1277–1286 CrossRef CAS PubMed.
- M. Seydou, G. Grégoire, J. Liquier, J. Lemaire, J. P. Schermann and C. Desfrançois, J. Am. Chem. Soc., 2008, 130, 4187–4195 CrossRef CAS PubMed.
- N. Takeda, K. Hirata, K. Tsuruta, G. D. Santis, X. Sotiris, S.-I. Ishiuchi and M. Fujii, Phys. Chem. Chem. Phys., 2021, 24, 5786–5793 RSC.
- G. Papadopoulos, A. Svendsen, O. V. Boyarkin and T. R. Rizzo, J. Am. Soc. Mass Spectrom., 2012, 23, 1173–1181 CrossRef CAS PubMed.
- M. Z. Kamrath and T. R. Rizzo, Acc. Chem. Res., 2018, 51, 1487–1495 CrossRef CAS PubMed.
- N. J. Coughlan, M. H. Stockett, C. Kjær, E. K. Ashworth, P. C. Bulman Page, S. R. Meech, S. Brøndsted Nielsen, L. Blancafort, W. S. Hopkins and J. N. Bull, J. Chem. Phys., 2021, 155, 124304 CrossRef CAS PubMed.
- F. V. Heldmaier, N. J. Coughlan, A. Haack, R. Huard, M. Guna, B. B. Schneider, J. Y. Le Blanc, J. L. Campbell, M. Nooijen and W. S. Hopkins, Phys. Chem. Chem. Phys., 2021, 23, 19892–19900 RSC.
- N. Mashmoushi, D. R. Juhász, N. J. Coughlan, B. B. Schneider, J. Y. Le Blanc, M. Guna, B. E. Ziegler, J. L. Campbell and W. S. Hopkins, J. Phys. Chem. A, 2021, 125, 8187–8195 CrossRef CAS PubMed.
- N. J. Coughlan, W. Fu, M. Guna, B. B. Schneider, J. Y. Le Blanc, J. L. Campbell and W. S. Hopkins, Phys. Chem. Chem. Phys., 2021, 23, 20607–20614 RSC.
- B. D. Adamson, N. J. Coughlan, G. da Silva and E. J. Bieske, J. Phys. Chem. A, 2013, 117, 13319–13325 CrossRef CAS PubMed.
- P. B. Markworth, B. D. Adamson, N. J. Coughlan, L. Goerigk and E. J. Bieske, Phys. Chem. Chem. Phys., 2015, 17, 25676–25688 RSC.
- M. S. Scholz, J. N. Bull, N. J. Coughlan, E. Carrascosa, B. D. Adamson and E. J. Bieske, J. Phys. Chem. A, 2017, 121, 6413–6419 CrossRef CAS PubMed.
- J. N. Bull, E. Carrascosa, N. Mallo, M. S. Scholz, G. Da Silva, J. E. Beves and E. J. Bieske, J. Phys. Chem. Lett., 2018, 9, 665–671 CrossRef CAS PubMed.
- J. N. Bull, J. T. Buntine, E. Carrascosa, M. H. Stockett and E. J. Bieske, Eur. Phys. J. D, 2021, 75, 1–12 CrossRef.
- U. Jacovella, E. Carrascosa, J. T. Buntine, N. Ree, K. V. Mikkelsen, M. Jevric, K. Moth-Poulsen and E. J. Bieske, J. Phys. Chem. Lett., 2020, 11, 6045–6050 CrossRef CAS PubMed.
- J. N. Bull, E. Carrascosa, L. Giacomozzi, E. J. Bieske and M. H. Stockett, Phys. Chem. Chem. Phys., 2018, 20, 19672–19681 RSC.
- C. Kjær, J. N. Bull, E. Carrascosa, S. B. Nielsen and E. J. Bieske, Eur. Phys. J. D, 2021, 75, 1–9 CrossRef.
- J. N. Bull, C. W. West, C. S. Anstöter, G. da Silva, E. J. Bieske and J. R. Verlet, Phys. Chem. Chem. Phys., 2019, 21, 10567–10579 RSC.
- J. N. Bull, N. J. Coughlan and E. J. Bieske, J. Phys. Chem. A, 2017, 121, 6021–6027 CrossRef CAS PubMed.
- J. N. Bull, J. T. Buntine, M. S. Scholz, E. Carrascosa, L. Giacomozzi, M. H. Stockett and E. J. Bieske, Faraday Discuss., 2019, 217, 34–46 RSC.
- J. N. Bull, M. S. Scholz, E. Carrascosa and E. J. Bieske, Phys. Chem. Chem. Phys., 2018, 20, 509–513 RSC.
- J. N. Bull, M. S. Scholz, E. Carrascosa, G. Da Silva and E. J. Bieske, Phys. Rev. Lett., 2018, 120, 223002 CrossRef CAS PubMed.
- J. N. Bull, G. d Silva, M. S. Scholz, E. Carrascosa and E. J. Bieske, J. Phys. Chem. A, 2019, 123, 4419–4430 CrossRef CAS PubMed.
- C. M. Choi, A.-L. Simon, F. Chirot, A. Kulesza, G. Knight, S. Daly, L. MacAleese, R. Antoine and P. Dugourd, J. Phys. Chem. B, 2016, 120, 709–714 CrossRef CAS PubMed.
- I. Czerwinska, A. Kulesza, C. Choi, F. Chirot, A.-L. Simon, J. Far, C. Kune, E. De Pauw and P. Dugourd, Phys. Chem. Chem. Phys., 2016, 18, 32331–32336 RSC.
- S. W. Smith, Toxicol. Sci., 2009, 110, 4–30 CrossRef CAS PubMed.
- H. Murashima and A. Fujihara, Chem. Phys., 2020, 536, 110818 CrossRef CAS.
- H. Murashima and A. Fujihara, Anal. Biochem., 2021, 114151 CrossRef CAS PubMed.
- H. J. Eun, A. Min, C. W. Jeon, I. T. Yoo, J. Heo and N. J. Kim, J. Phys. Chem. Lett., 2020, 11, 4367–4371 CrossRef CAS PubMed.
- I. T. Yoo, H. J. Eun, A. Min, C. W. Jeon, J. Jeong, J. Heo and N. J. Kim, Phys. Chem. Chem. Phys., 2021, 23, 24180–24186 RSC.
- S. Daly, F. Rosu and V. Gabelica, Science, 2020, 368, 1465–1468 CrossRef CAS PubMed.
- P. Krüger and K.-M. Weitzel, Angew. Chem., 2021, 60, 17861–17865 CrossRef PubMed.
- V. Gabelica, Acc. Chem. Res., 2021, 54, 3691–3699 CrossRef CAS PubMed.
- M. F. Matus, S. Malola, E. K. Bonilla, B. M. Barngrover, C. M. Aikens and H. Häkkinen, Chem. Commun., 2020, 56, 8087–8090 RSC.
- E. Kalenius, S. Malola, M. F. Matus, R. Kazan, T. Bürgi and H. Häkkinen, J. Am. Chem. Soc., 2021, 143, 1273–1277 CrossRef CAS PubMed.
- A. Baksi, P. Chakraborty, S. Bhat, G. Natarajan and T. Pradeep, Chem. Commun., 2016, 52, 8397–8400 RSC.
- A. Baksi, E. K. Schneider, P. Weis, K. Krishnadas, D. Ghosh, H. Hahn, T. Pradeep and M. M. Kappes, J. Phys. Chem. C, 2019, 123, 28477–28485 CrossRef CAS.
- K. Hirata, K. Yamashita, S. Muramatsu, S. Takano, K. Ohshimo, T. Azuma, R. Nakanishi, T. Nagata, S. Yamazoe and K. Koyasu, Nanoscale, 2017, 9, 13409–13412 RSC.
- K. Hirata, K. Kim, K. Nakamura, H. Kitazawa, S. Hayashi, K. Koyasu and T. Tsukuda, J. Phys. Chem. C, 2019, 123, 13174–13179 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |