
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxamic acids: useful precursors of carbamoyl radicals
Ikechukwu Martin
Ogbu
ab,
Gülbin
Kurtay
 ac,
Frédéric
Robert
a and
Yannick
Landais
*a
ac,
Frédéric
Robert
a and
Yannick
Landais
*a
aUniversity of Bordeaux, Institute of Molecular Sciences (ISM), UMR-CNRS 5255, 351, Cours de la Libération, 33405 Talence, Cedex, France. E-mail: yannick.landais@u-bordeaux.fr
bAlex Ekwueme Federal University, Department of Chemistry, Faculty of Sciences, Ndufu-Alike Ikwo, Abakaliki, Ebonyi State, Nigeria
cUniversity of Ankara, Department of Chemistry, Faculty of Science, Ankara, Turkey
First published on 16th June 2022
Abstract
This review article describes the recent development in the chemistry of carbamoyl radicals generated from oxamic acids. This mild and efficient method compares well with previous methods of generation of these nucleophilic radicals. The oxidative decarboxylation of oxamic acids can be mediated through thermal, photochemical, electrochemical or photoelectrochemical means, generating carbamoyl radicals, which may further add to unsaturated systems to provide a broad range of important amides. Oxidative decarboxylation of oxamic acids also offers a straightforward entry for the preparation of urethanes, ureas, and thioureas.
 Ikechukwu Martin Ogbu | Ikechukwu Martin Ogbu received his MSc in pure and industrial chemistry from Nnamdi Azikiwe University, Nigeria, and a PhD in organic chemistry from the University of Bordeaux, France, under the direction of Professor Yannick Landais. His work focused mainly on the development of new processes for urethanes and urea synthesis. He is currently a lecturer at Alex Ekwueme Federal University Ndufu-Alike, Nigeria. |
 Gülbin Kurtay | Gülbin Kurtay received her PhD in Chemistry from Ankara University (Turkey), where she was promoted to Assistant Professor (2020). She is currently a postdoctoral researcher as a recipient of the TUBITAK BIDEB 2219-International Postdoctoral Research Fellowship at Bordeaux University (France) under the supervision of Prof. Yannick Landais. Her current research interests are in the fields of photochemical and electrochemical transformation of oxamic acid derivatives. |
 Frédéric Robert | Frédéric Robert obtained his PhD degree in 1999 from Grenoble University under the supervision of Dr Andrew E. Greene and Dr Yves Gimbert. He moved to Dartmouth College (New Hampshire, USA) in 2000 for a postdoctoral stay with Prof. Peter A. Jacobi, followed by a second postdoctoral stay in 2001 at the University of Geneva (CH) with Prof. Peter Kündig. He was appointed CNRS research associate at the University of Bordeaux in 2002. His current research focuses on mechanistic studies on radical chemistry using DFT. |
 Yannick Landais | Yannick Landais received his PhD degree from the University of Orsay (Paris XI) under the supervision of Dr Jean-Pierre Robin. After a post-doctoral work with Prof. Ian Fleming at Cambridge University, he joined the University of Lausanne as an assistant-Professor and was then appointed at the University of Bordeaux, where he is currently Professor of organic chemistry. His research interests are in radical and organosilicon chemistry, and application in natural product synthesis and polymer chemistry. He is a recipient of several awards including the Werner prize of the New Swiss Chemical Society and the Prize of the Organic Division of the French Chemical Society. |
1. Introduction
Oxamic acids I also known to as oxalic acid monoamides have emerged as potent precursors for the generation of the carbamoyl radical II (Fig. 1). Oxamic acids can easily undergo decarboxylation through a single electron oxidation resulting in the generation of the reactive carbamoyl radical, which can then engage in diverse radical reactions or undergo a second single electron oxidation as originally unveiled by Minisci.1 Oxamic acids are thus versatile intermediates for the synthesis of nitrogen-containing organic molecules.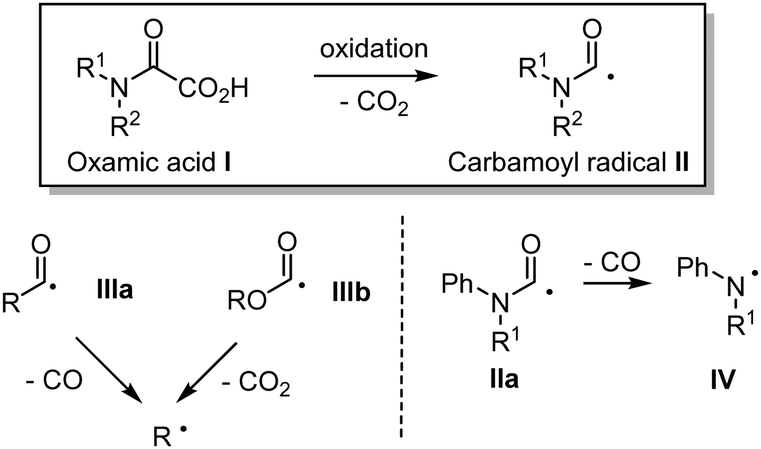 | ||
| Fig. 1 Carbamoyl radicals from oxamic acids. Related acyl radicals. | ||
Ekad and Rokach first demonstrated that carbamoyl radical II, generated through photolysis of formamides (R2NC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)H) was able to add to terminal and non-terminal olefins.2II is nucleophilic in nature and therefore readily adds to electron deficient systems, but also to alkynes, arenes, and heteroarenes, providing a reliable tool for the construction of nitrogen-containing substrates.3 Carbamoyl radicals may be observed by Electron-Spin-Resonance (ESR) depending on the nature of the substitution on the nitrogen.4 For instance, N-alkyl and N-alkyl-N-aryl carbamoyl radicals are both stable enough to be observed through ESR,5 while the N-arylcarbamoyl analogue IIa is transient and tends to decarbonylate to generate the more stable persistent arylaminyl radical IV. Sutcliffe and Ingold6 showed through ESR and NMR experiments that the relative proportion of the s-cis and s-trans conformations of the carbamoyl radical II, resulting from the abstraction of formamides CHO hydrogen paralleled that of the parent formamide conformations in every respect. Radical II is a σ-type radical, which displays a reactivity similar to that of closely related α-type acyl IIIa and alkoxycarbonyl IIIb radicals (Fig. 1).7 However and in contrast, radical II lifetime is relatively long as compared to that of IIIa and IIIb, which are prone to decarbonylation and decarboxylation respectively, resulting in the generation of the more stable alkyl radicals.8 Decarbonylation of carbamoyl radical II is less favoured (except in arylaminyl cases IIa, vide supra), as it would lead to a higher energetic aminyl radical. Although less attention was paid to carbamoyl radical as compared to its acyl radical analogues, recent years have witnessed an increasing interest in the use of II in synthetic organic chemistry, which may be attributed among other factors, to the renaissance of oxamic acid as its potent precursor. Carbamoyl radical is traditionally generated by the homolytic cleavage of R2NC(O)–X precursors using a radical initiator, heat, or UV light, where X can be H,9,10 SPh,11 xanthate,12 Co(salophen),7 cyclohexadienyl,13 TePh,14 SePh15 or Cl.16 Other methods to access carbamoyl radicals include photocatalyzed cleavage of weak S–C bond in dithiocarbamate,17a or carbamoyl-Hantzsch esters,17b–e reductive decarboxylation of N-hydroxyphthalimido oxamides,18 and oxidation of phenylhydrazinecarboxamide.19 These strategies have their own merits but often rely on the synthesis of precursors, which require several steps and the use of complex reagents, leading to poor atom economy. Regioselectivity problems were also observed, for instance during the generation of the carbamoyl radical through formamides C–H abstraction, which can provide a mixture of products, resulting from the competitive abstraction at CHO and α to nitrogen.9,20 In contrast, the generation of carbamoyl radicals through decarboxylation of oxamic acids has been shown to be very efficient and circumvent the regioselectivity issues associated with classical routes. Unlike the well-studied decarboxylative coupling of carboxylic acids and ketoacids, decarboxylation of oxamic acids proceeds under mild reaction conditions. Oxamic acids are bench stable and non-toxic compounds that can be easily prepared by direct coupling of amines with commercially available oxalic acid monoester derivatives (Scheme 1).21 The oxamate ester intermediate may be hydrolysed under basic conditions when methyl and ethyl esters are present, while acidic conditions were found very convenient with t-Bu esters directly leading to the oxamic acid. This simple two-step procedure gives access to oxamic acids of wide structural diversity using various amines and does not require tedious purification processes. We describe in this review article recent efforts to exploit the versatile reactivity of the simple, yet attractive, carbamoyl radical in addition, coupling and oxidative processes.
O)H) was able to add to terminal and non-terminal olefins.2II is nucleophilic in nature and therefore readily adds to electron deficient systems, but also to alkynes, arenes, and heteroarenes, providing a reliable tool for the construction of nitrogen-containing substrates.3 Carbamoyl radicals may be observed by Electron-Spin-Resonance (ESR) depending on the nature of the substitution on the nitrogen.4 For instance, N-alkyl and N-alkyl-N-aryl carbamoyl radicals are both stable enough to be observed through ESR,5 while the N-arylcarbamoyl analogue IIa is transient and tends to decarbonylate to generate the more stable persistent arylaminyl radical IV. Sutcliffe and Ingold6 showed through ESR and NMR experiments that the relative proportion of the s-cis and s-trans conformations of the carbamoyl radical II, resulting from the abstraction of formamides CHO hydrogen paralleled that of the parent formamide conformations in every respect. Radical II is a σ-type radical, which displays a reactivity similar to that of closely related α-type acyl IIIa and alkoxycarbonyl IIIb radicals (Fig. 1).7 However and in contrast, radical II lifetime is relatively long as compared to that of IIIa and IIIb, which are prone to decarbonylation and decarboxylation respectively, resulting in the generation of the more stable alkyl radicals.8 Decarbonylation of carbamoyl radical II is less favoured (except in arylaminyl cases IIa, vide supra), as it would lead to a higher energetic aminyl radical. Although less attention was paid to carbamoyl radical as compared to its acyl radical analogues, recent years have witnessed an increasing interest in the use of II in synthetic organic chemistry, which may be attributed among other factors, to the renaissance of oxamic acid as its potent precursor. Carbamoyl radical is traditionally generated by the homolytic cleavage of R2NC(O)–X precursors using a radical initiator, heat, or UV light, where X can be H,9,10 SPh,11 xanthate,12 Co(salophen),7 cyclohexadienyl,13 TePh,14 SePh15 or Cl.16 Other methods to access carbamoyl radicals include photocatalyzed cleavage of weak S–C bond in dithiocarbamate,17a or carbamoyl-Hantzsch esters,17b–e reductive decarboxylation of N-hydroxyphthalimido oxamides,18 and oxidation of phenylhydrazinecarboxamide.19 These strategies have their own merits but often rely on the synthesis of precursors, which require several steps and the use of complex reagents, leading to poor atom economy. Regioselectivity problems were also observed, for instance during the generation of the carbamoyl radical through formamides C–H abstraction, which can provide a mixture of products, resulting from the competitive abstraction at CHO and α to nitrogen.9,20 In contrast, the generation of carbamoyl radicals through decarboxylation of oxamic acids has been shown to be very efficient and circumvent the regioselectivity issues associated with classical routes. Unlike the well-studied decarboxylative coupling of carboxylic acids and ketoacids, decarboxylation of oxamic acids proceeds under mild reaction conditions. Oxamic acids are bench stable and non-toxic compounds that can be easily prepared by direct coupling of amines with commercially available oxalic acid monoester derivatives (Scheme 1).21 The oxamate ester intermediate may be hydrolysed under basic conditions when methyl and ethyl esters are present, while acidic conditions were found very convenient with t-Bu esters directly leading to the oxamic acid. This simple two-step procedure gives access to oxamic acids of wide structural diversity using various amines and does not require tedious purification processes. We describe in this review article recent efforts to exploit the versatile reactivity of the simple, yet attractive, carbamoyl radical in addition, coupling and oxidative processes.
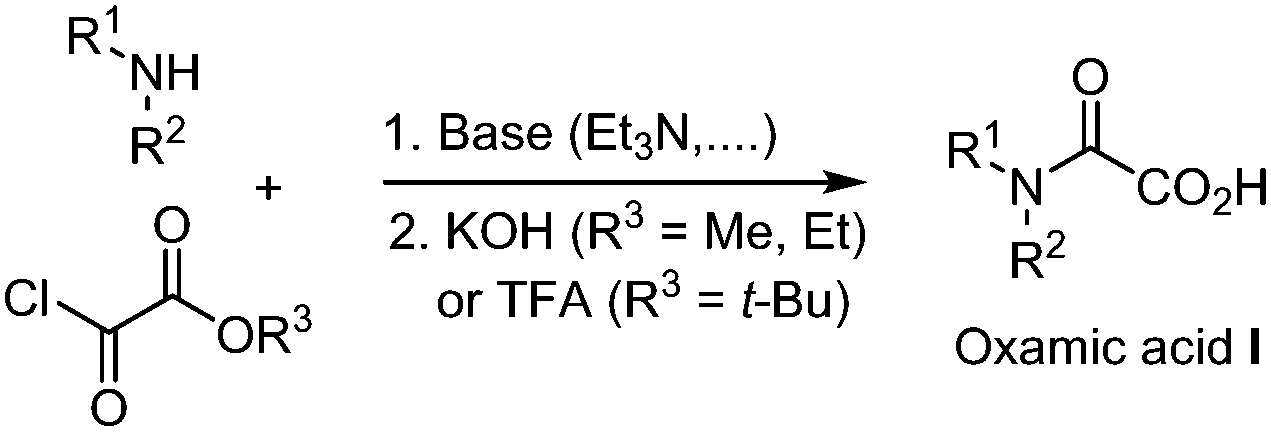 | ||
| Scheme 1 Oxamic acid preparation from oxalic acid chloride monoester. | ||
2. Decarboxylative coupling reactions using oxamic acids
As mentioned earlier, Minisci and co-workers1,9a,b originally showed that oxidative decarboxylation of oxamic acids 1 using a stoichiometric amount of ammonium persulfate as oxidant and a silver salt as a catalyst generates the carbamoyl radical II, which could then be trapped by a protonated heteroaromatic base 2,22 to furnish the corresponding amide 3 in excellent yield (Scheme 2). According to their mechanism, the Ag(II) species, generated through the oxidation of Ag(I) by the persulfate, triggered the oxidation of oxamic acid into carbamoyl radical II. This seminal work has continued to attract increasing attention in organic synthesis and has led to many different simple procedures for the preparation of amides of biological interests using oxamic acids.23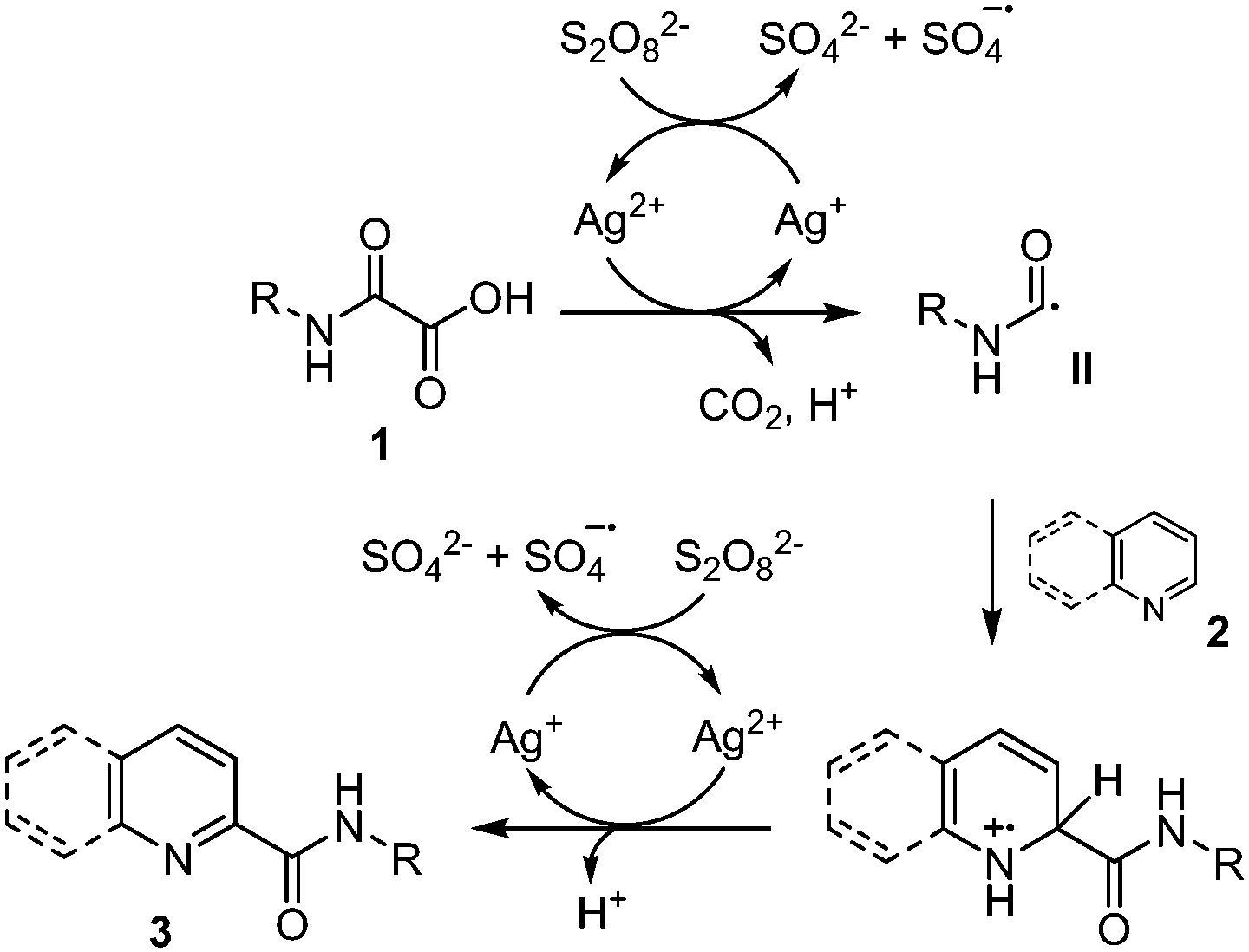 | ||
| Scheme 2 Ag(I)/persulfate-mediated generation of carbamoyl radical II and addition to heteroarenes. | ||
A silver-free version of the above Minisci reaction was recently reported by different laboratories and demonstrated to be efficient for carbamoylation of a variety of heteroarenes, including quinoxalin-2(1H)-ones,24 phenanthrolines25 and 2H-indazoles (Scheme 3).26 This procedure relies on the use of (NH4)2S2O8 as an oxidant, DMSO/H2O as solvents, and a temperature between 40–60 °C to furnish the corresponding amides 3 in moderate to high yield, without the need for a silver salt catalyst or acidification. The choice of solvent proved to be crucial, the reaction being greatly accelerated in a DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (600:1) mixture. DMSO or water alone gave inferior product yield, while other organic solvents such as DCE, CH3CN, MeOH, and 1,4-dioxane were not suitable for the reaction. The procedure led to high yield of amidation of quinolines (Scheme 3, (a))25a quinoxalin-2(1H)-ones, (b)24 and 2H-indazoles, (c).26 A direct C–H functionalization of phenanthrolines, useful ligands in Cu- or Fe-catalyzed processes, has also been carried out using this strategy, (d).25b Polyamides were thus at hand through a simple treatment of phenanthroline precursors with excess oxamic acid.
:H2O (600:1) mixture. DMSO or water alone gave inferior product yield, while other organic solvents such as DCE, CH3CN, MeOH, and 1,4-dioxane were not suitable for the reaction. The procedure led to high yield of amidation of quinolines (Scheme 3, (a))25a quinoxalin-2(1H)-ones, (b)24 and 2H-indazoles, (c).26 A direct C–H functionalization of phenanthrolines, useful ligands in Cu- or Fe-catalyzed processes, has also been carried out using this strategy, (d).25b Polyamides were thus at hand through a simple treatment of phenanthroline precursors with excess oxamic acid.
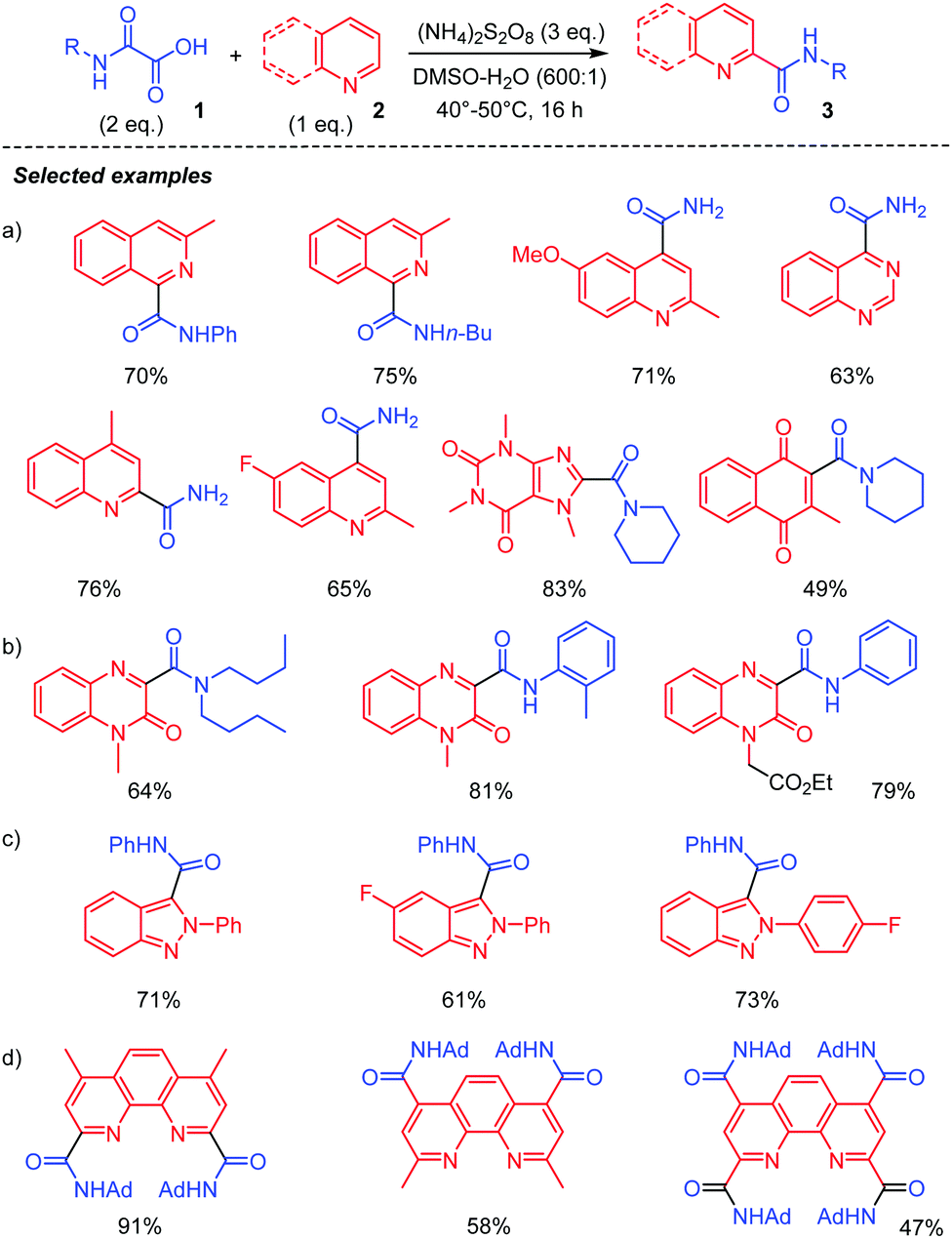 | ||
| Scheme 3 Persulfate-mediated generation of carbamoyl radical II and addition to heteroarenes. | ||
A plausible mechanism proposed by Yuan and co-workers24 (Scheme 4) suggests that the reaction proceeds first by homolytic cleavage of S2O82− in DMSO to give SO4˙−, the latter then mediating the oxidative decarboxylation of oxamic acid 1 into carbamoyl radical II. A regioselective addition of the carbamoyl radical onto the heteroarene 2 would then follow, leading to intermediate i. A single electron transfer (SET) from i mediated by S2O82− would then give a cationic species, which undergoes deprotonation and rearomatization to afford the final product 3.
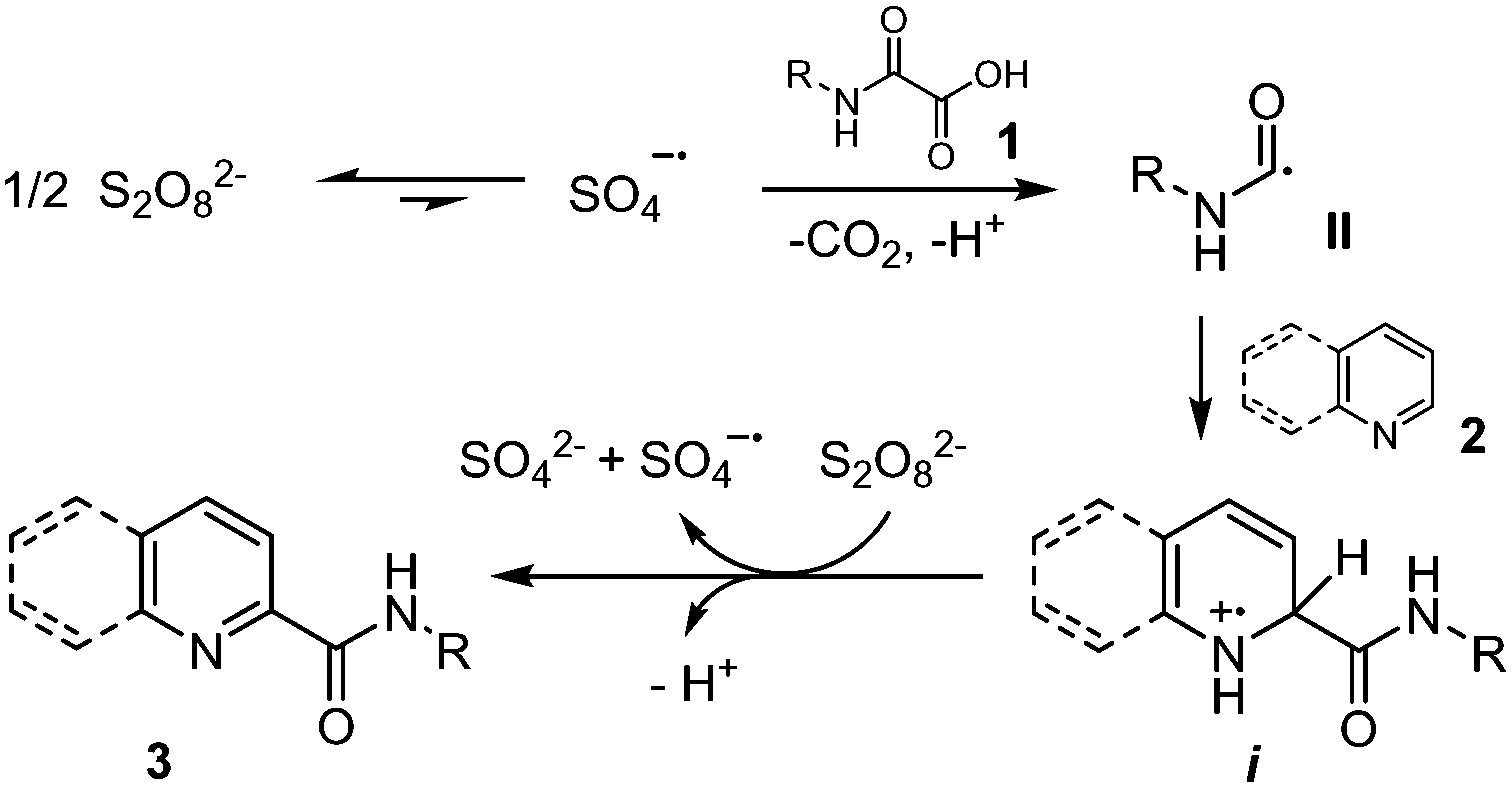 | ||
| Scheme 4 Mechanism of the metal-free persulfate decarboxylation of oxamic acids and addition to heteroarenes. | ||
Our group recently developed a visible light-mediated carbamoyl radical addition to heteroarenes using oxamic acids (Scheme 5).27 This metal-free and efficient process operates at room temperature, using a hypervalent iodine reagent, i.e. acetoxybenziodoxolone (BI-OAc) as an oxidant, and an organic dye, i.e. 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4-CzIPN) to mediate the oxidative decarboxylation of oxamic acid 1 into the carbamoyl radical II, which in the presence of heteroaromatic base 2 delivered the corresponding amide 3 in good to high yields. The procedure is compatible with a wide range of oxamic acids 1, including chiral ones to furnish the desired chiral amides without racemization. The involvement of the carbamoyl radical II was unambiguously demonstrated through its trapping with (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), an alkynylsulfone, or an allylsilane, leading respectively to a TEMPO-II adduct, an alkynylamide, or an unsaturated amide. A plausible mechanism for the transformation suggests that the photoexcited catalyst (4-CzIPN*) is quenched by a hypoiodite species i formed by a ligand exchange between oxamic acid 1 and BI-OAc (Scheme 5). The generated unstable radical anion ii collapses to give the carbamoyl radical II. The latter then undergoes radical addition to the heteroaromatic base 2 to give a radical intermediate iii, which is oxidized by the photocatalyst in its semi-oxidized form (4-CzIPN·+) to give cationic species iv. Deprotonation and rearomatization of iv then furnish the product 3. As proposed by Minisci,1 the carbamoyl radical addition likely occurs onto the protonated heteroaromatic base 2 under these conditions where acetic acid is produced in the first step. Very recently, Chen, Yu, and co-workers28 repeated the above experiments using a heterogeneous conjugated polymeric photocatalyst (CPP3) based on 4-CzIPN and were able to catalyze the addition of a cyclohexyloxamic acid to quinoline albeit with a lower yield than with the simple 4-CzIPN (60% vs. 80% with 4-CzIPN, Scheme 5).
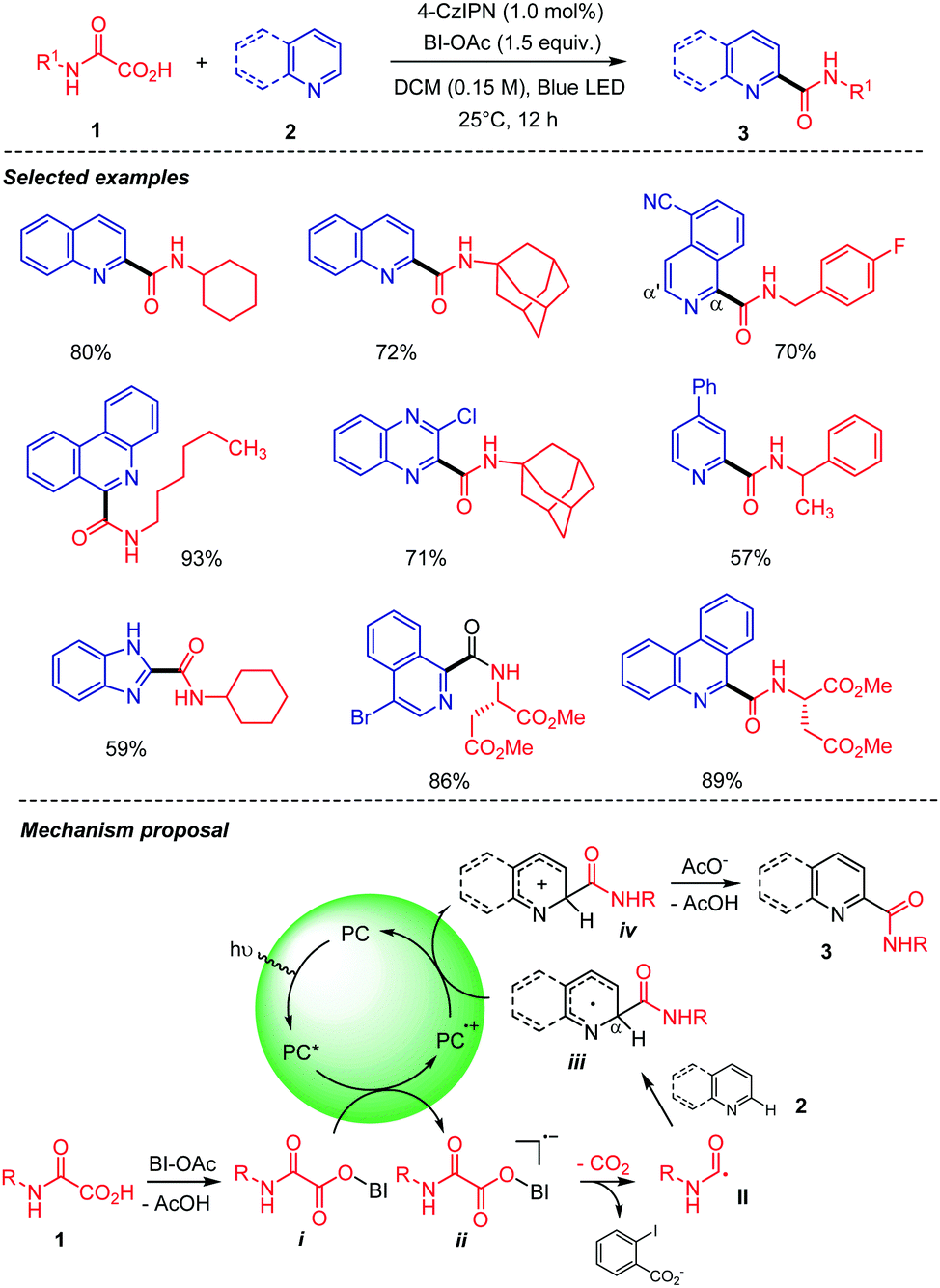 | ||
| Scheme 5 Photocatalyzed decarboxylation of oxamic acids in the presence of heteroarenes. Heteroarenes scope and mechanistic proposal. | ||
Jouffroy and Kong,29 in the meantime, reported a similar visible light mediated decarboxylative carbamoylation of heteroaromatic bases 5 using oxamic acids and their potassium oxamate 4 (Scheme 6). Their method employs potassium persulfate as an oxidant in aqueous DMSO to achieve the decarboxylative carbamoylation under visible light irradiation using an acridinium-based photocatalyst. The desired amides 6 were obtained in moderate to good yields using aliphatic potassium oxamates. Interestingly, from a mechanistic point of view, the photocatalyst in its excited state is reduced by the oxamate salt and the oxidant is used here to reoxidize the photocatalyst back to its ground state. The reaction led to good results whatever the primary or secondary nature of the oxamic acid.
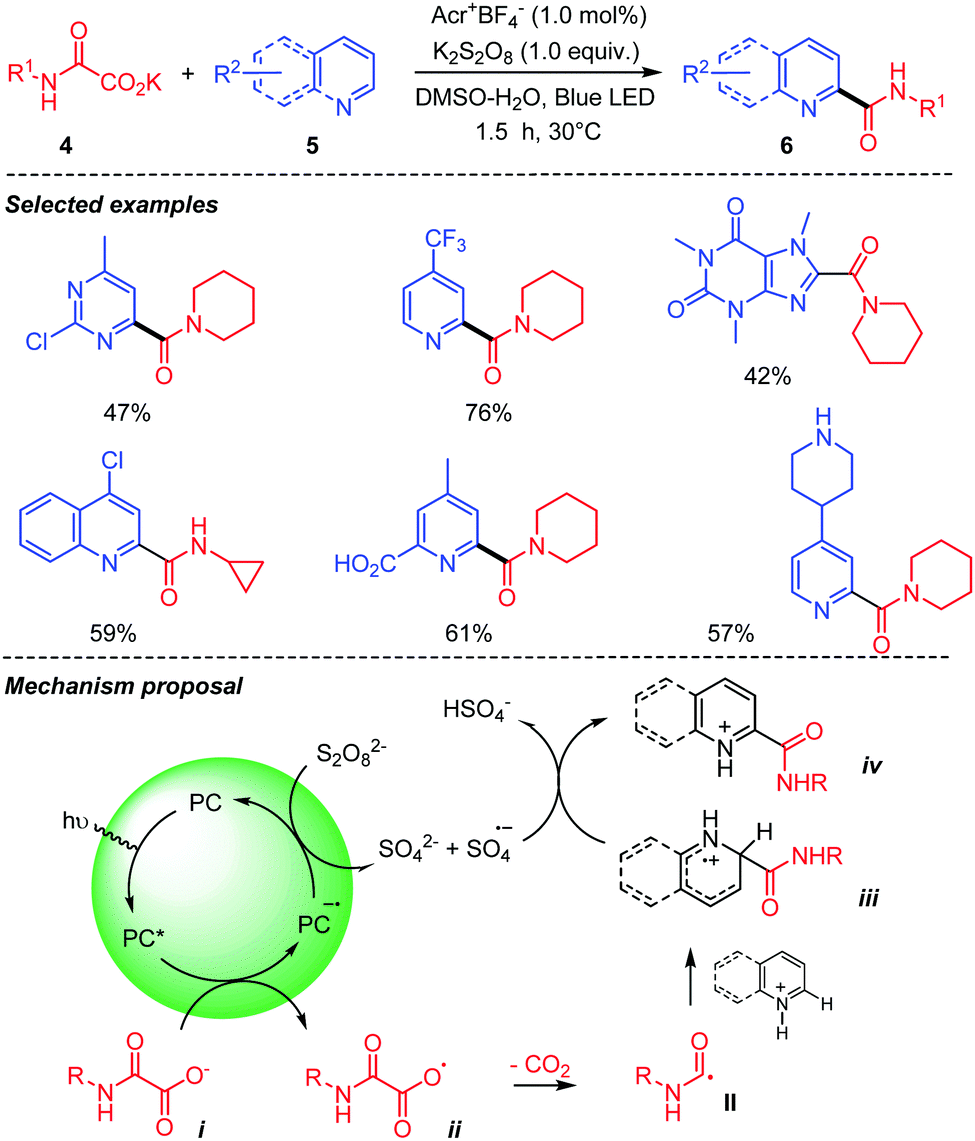 | ||
| Scheme 6 Acridinium-mediated photocatalyzed decarboxylation of oxamic acids in the presence of heteroarenes. | ||
In 2020, Song and co-workers30 unveiled an electrophotocatalytic decarboxylative Minisci reaction between oxamic acids 7 and heteroarenes 8 (Scheme 7). This strategy, that combined organic electrochemistry, and photocatalysis with the anodic oxidation replacing the chemical oxidant, was shown to be efficient for the preparation of various amides 9. The authors demonstrated that both the light and electricity were essential for the transformation as only 3 to 18% of the desired product was observed in the absence of either of them. The procedure was compatible with a wide range of oxamic acids and heteroarenes with good functional group tolerance. A plausible mechanism for the transformation involves a single electron oxidative decarboxylation of the oxamate ion (Ep/2ox = +1.17 V vs. SCE) by the photoexcited catalyst 4-CzIPN* (Ep/2red = +1.35 V vs. SCE), followed by the addition of the generated carbamoyl radical II to the protonated heteroarene, to give a radical-cation intermediate i. The latter accepts an electron from the highly reducing photocatalyst radical anion (4-CzIPN·−) leading to intermediate ii, along with the regenerated photocatalyst. Finally, anodic oxidation of ii furnished the final product iii.
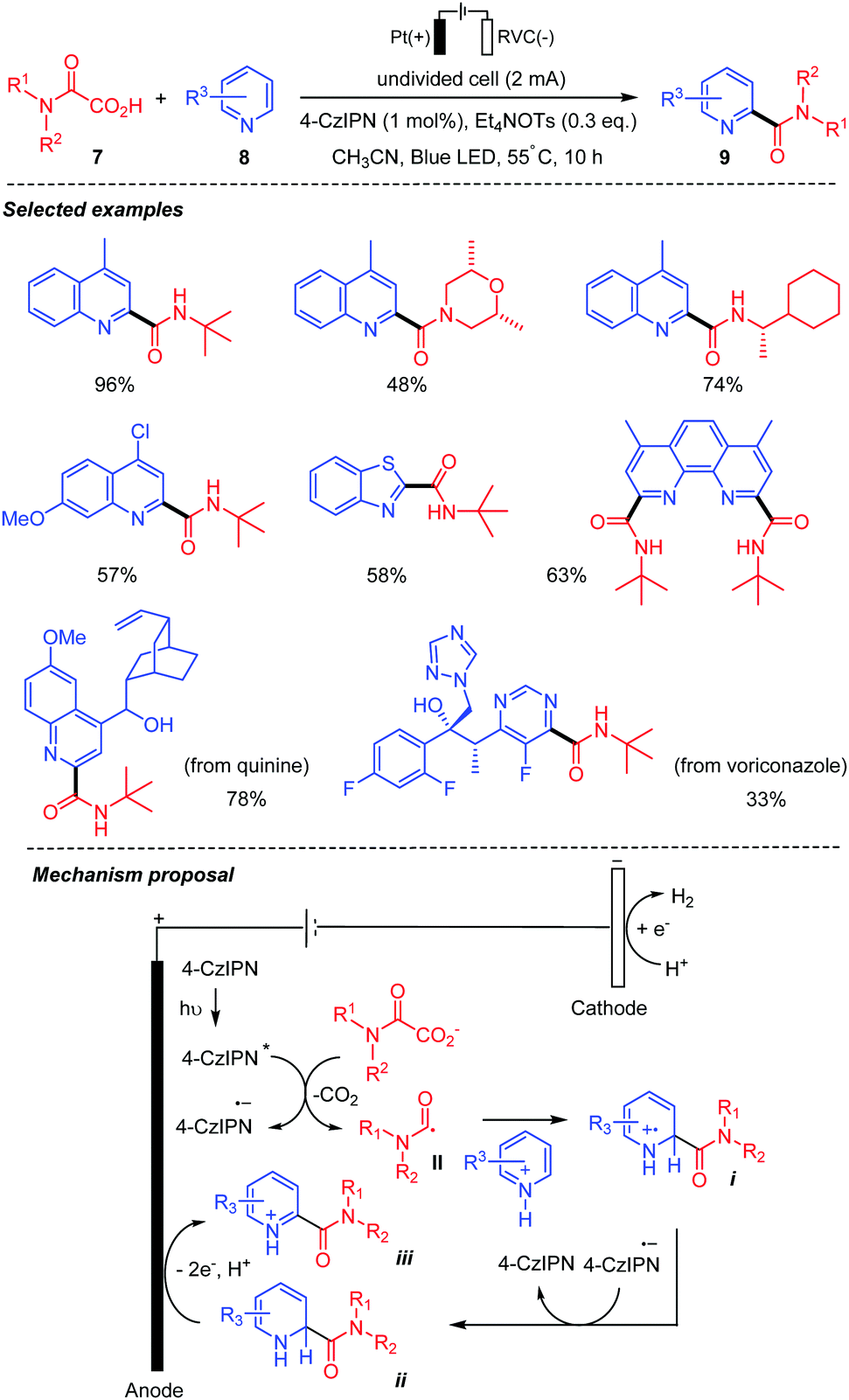 | ||
| Scheme 7 Electrophotocatalytic decarboxylative Minisci reaction between oxamic acids and heteroarenes. | ||
Besides heteroarenes as coupling partners, several other compounds can serve as competent coupling partners with oxamic acids, providing useful and efficient synthetic methods for the synthesis of amides. Wang and co-workers thus presented a palladium-catalyzed decarboxylative amidation using oxamic acids and arenes with ketoxime group 10 as a coupling partner (Scheme 8).31 The reaction works well with a wide range of N,N-disubstituted oxamic acids including cyclic ones to give the corresponding ortho-amidated ketoximes 11 in good yields and regioselectivity. With N-monosubstituted oxamic acids (7, R2 = H), isoindolinones 12 were obtained instead through an intramolecular cyclization of the ortho-amidated ketoximes formed. The authors proposed that the reaction proceeds through the formation of a five-membered palladacycle i (Scheme 8). This is followed by the oxidative addition of the carbamoyl radical II, issued from oxamic acid decarboxylation, to the complex, to form a Pd(IV) intermediate ii. The reductive elimination of ii would furnish product 11 and regenerates the Pd(II). When monosubstituted oxamic acid is used, the ortho-amidated ketoxime formed can further complex with Pd(II) to give a new complex iii, which undergoes intramolecular insertion to give intermediate iv. Then, removal of methoxyamine via β-H elimination affords the 3-methyleneisoindolinone 12.
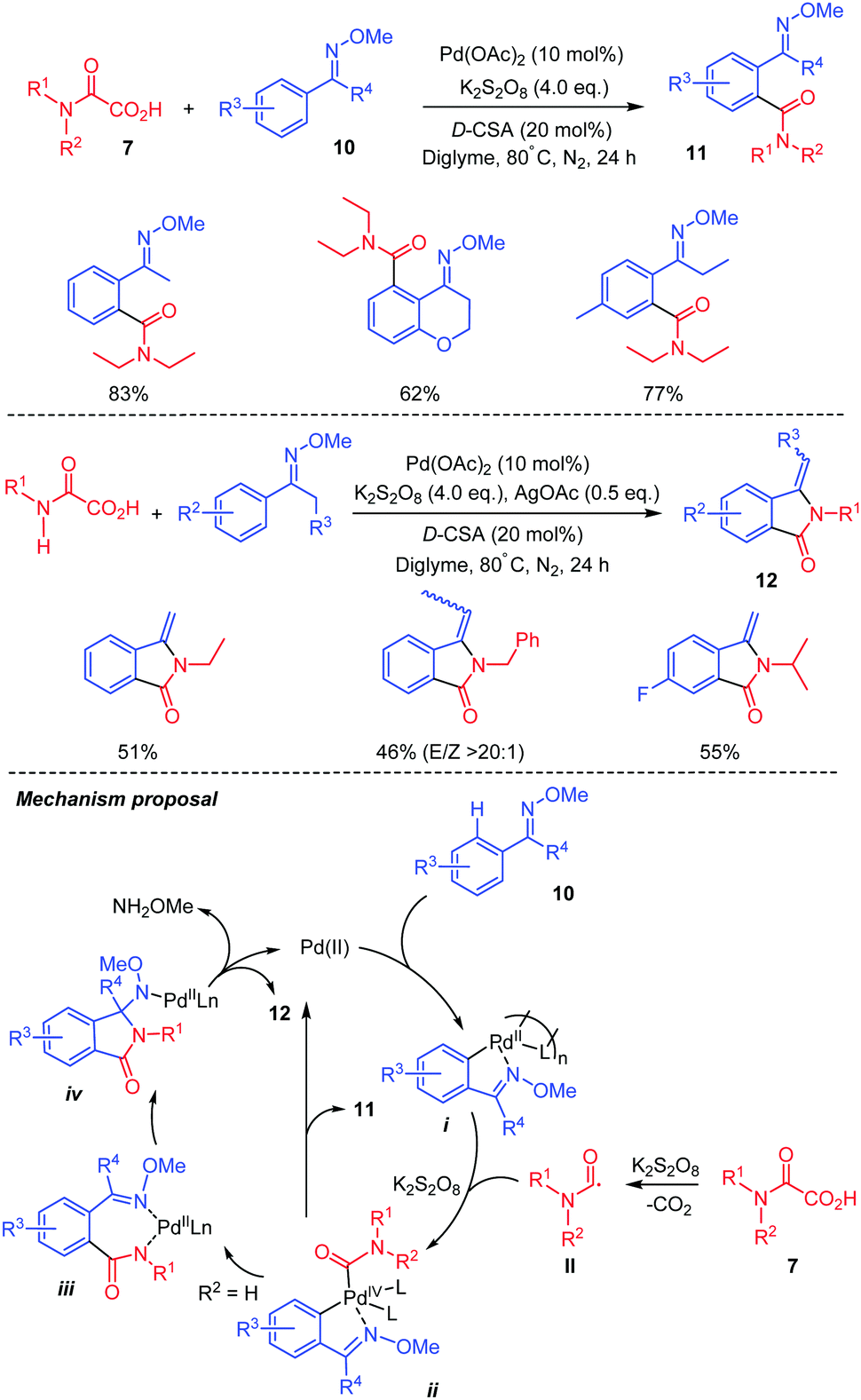 | ||
| Scheme 8 Pd(II)-catalyzed C–H amidation by oxamic acids, of arenes having a ketoxime directing group. | ||
Fu and co-workers32 reported a decarboxylative coupling of oxamic acids 7 and aryl halides 13 using a dual iridium and palladium photoredox catalytic system (Scheme 9). The authors showed that both catalytic partners were necessary for the reaction, as the desired product was not observed in the absence of either one of them. Although different possible mechanistic pathways were proposed for this transformation, in all, the photoexcited iridium (Ir(III)*) is implicated in the decarboxylation of the oxamic acid into carbamoyl radical II. Amongst three calculated pathways, one emerges and is depicted in Scheme 9 below, in which oxidative addition of Ar–X 13 onto Pd(0) provides a Pd(II) intermediate i, which is able to capture the carbamoyl radical II to generate a Pd(III) species ii. Reduction of the latter by Ir(II) restores Ir(III) in its ground state and a Pd(II) intermediate iv, the reductive elimination of which affords product 14 and Pd(0).
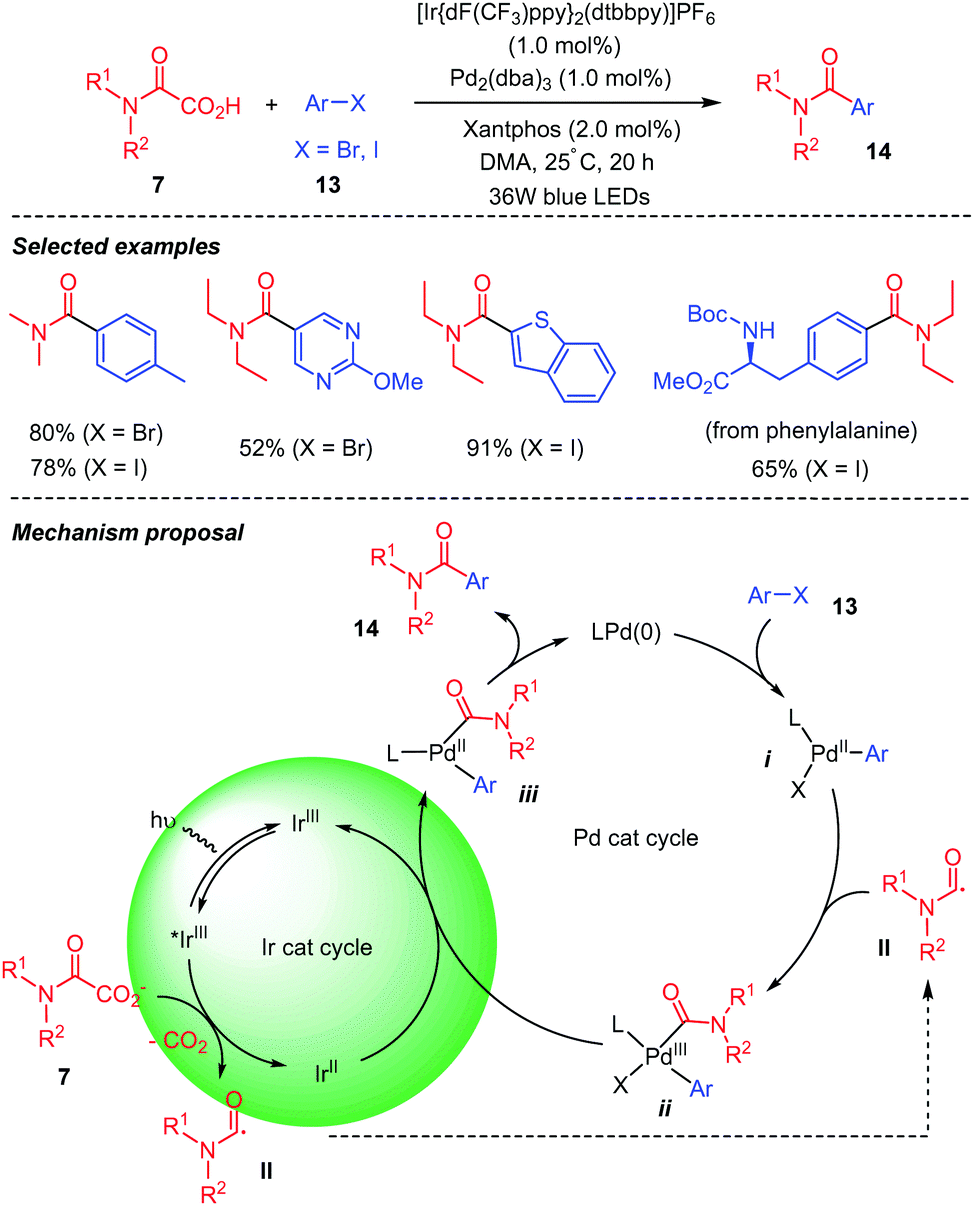 | ||
| Scheme 9 Dual Ir–Pd photoredox catalyzed decarboxylative cross-coupling of oxamic acids and aryl halides. | ||
Coupling of oxamic acids 7 with C(sp2) centers in cinnamic acids 15 has also been reported (Scheme 10).33 Fe(II) mediated oxidative decarboxylative cross-coupling of oxamic acids and acrylic acids was thus shown to afford α,β-unsaturated amides 16 (Scheme 10), albeit in moderate yields over a small number of examples. The mechanism is thought to proceed through the addition of the carbamoyl radical II onto the C(sp2) center bearing the CO2H group to generate a benzylic radical intermediate i. β-Elimination and release of CO2 from i afford the cinnamoyl amide 16. The persulfate oxidizes Fe(II) into Fe(III), the latter mediating the oxidative decarboxylation of the oxamic acid and the generation of the carbamoyl radical II.
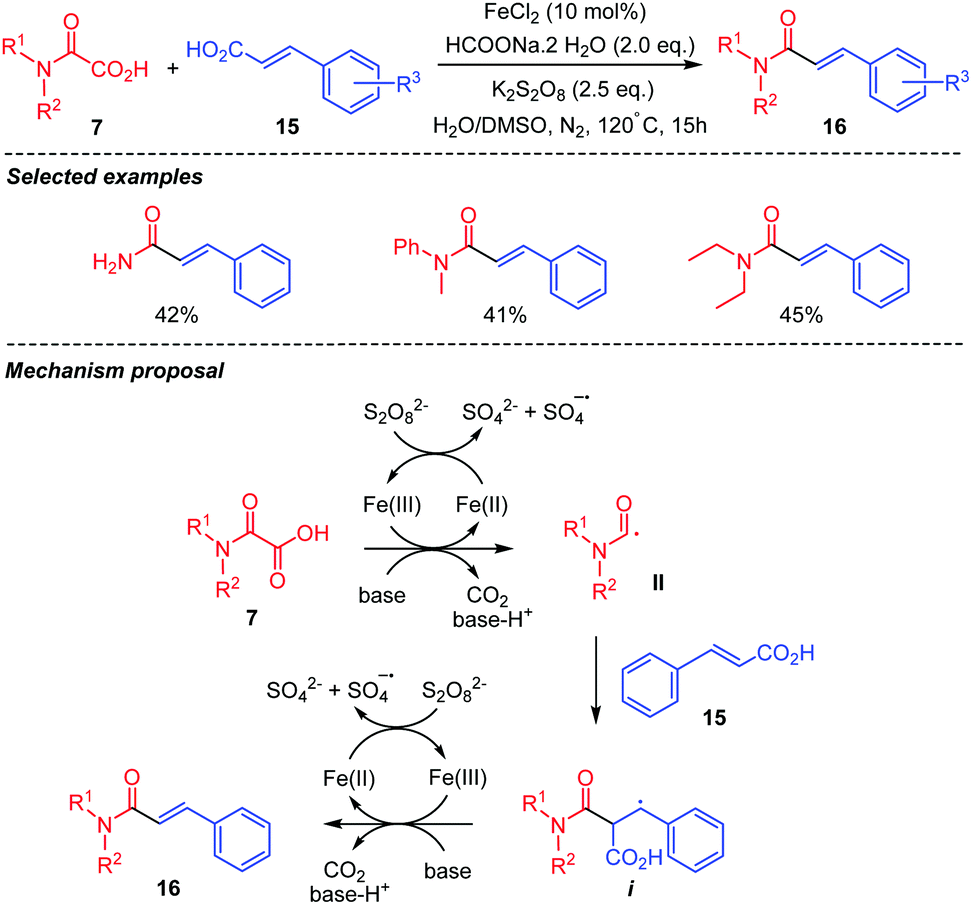 | ||
| Scheme 10 Fe(II)-mediated oxidative decarboxylative cross-coupling between oxamic acids and cinnamic acids. | ||
Oxamic acids can also be used for the formation of Csp2–Csp bonds through decarboxylative coupling with alkynyl compounds. In 2015, Duan's group presented a decarboxylative alkynylation of oxamic acids 7 using hypervalent alkynyl iodide reagent 17 as the alkynylating agent (Scheme 11).34 This strategy gave access to a wide range of propiolamides 18 in moderate yields using different oxamic acids and alkynyl coupling partners. Propiolamides are important synthons for organic synthesis and interesting building blocks for natural product synthesis.34 The authors proposed that the reaction proceeds through the K2S2O8 mediated decarboxylation of the oxamic acid 7 to generate carbamoyl radical II, which adds to the triple bond of the hypervalent iodine species 17, leading to radical intermediate i capable of undergoing a β-elimination to deliver the desired product 18 and the benziodoxolonyl radical ii. The latter is converted into benzoic acid through a reduction/protonation process.
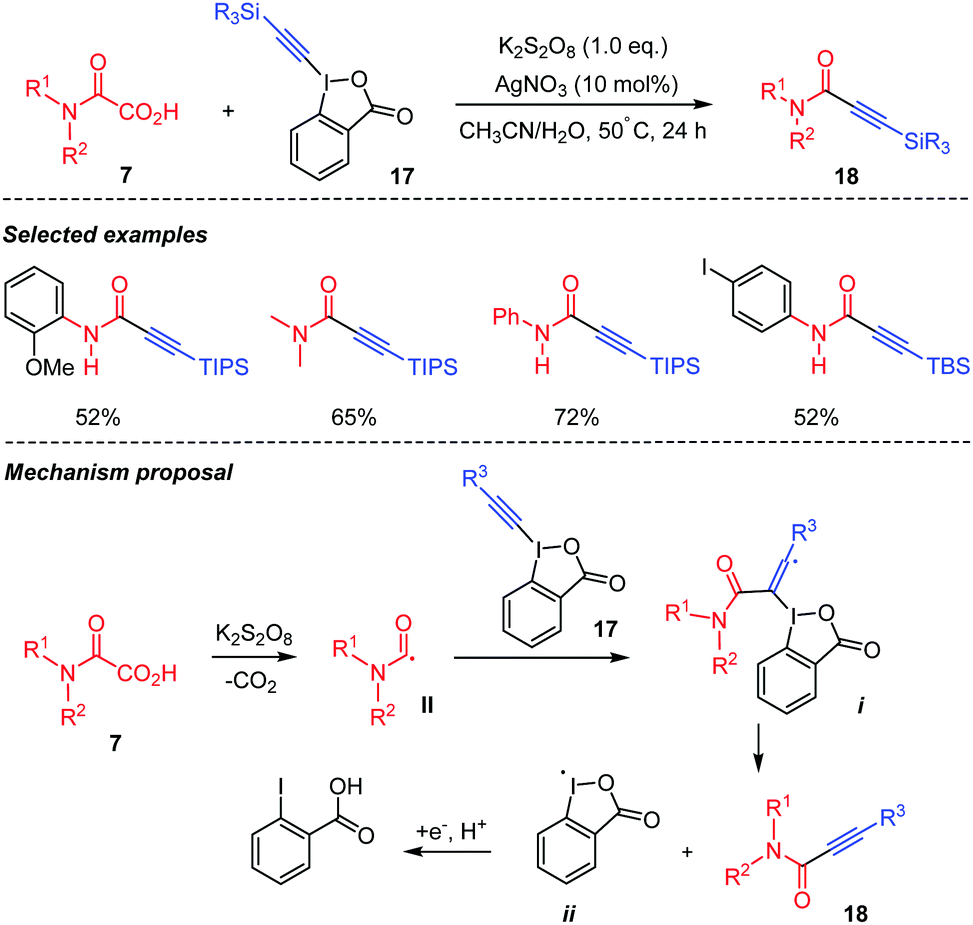 | ||
| Scheme 11 Decarboxylative alkynylation of oxamic acids. | ||
Chen's group proposed in the meantime a visible light mediated version of this decarboxylative coupling of oxamic acids with hypervalent alkynyl iodine species 19 for the preparation of ynamides 20 (Scheme 12).35 They demonstrated that the reaction was compatible with a wide range of oxamic acids, furnishing the corresponding ynamides in high yields.
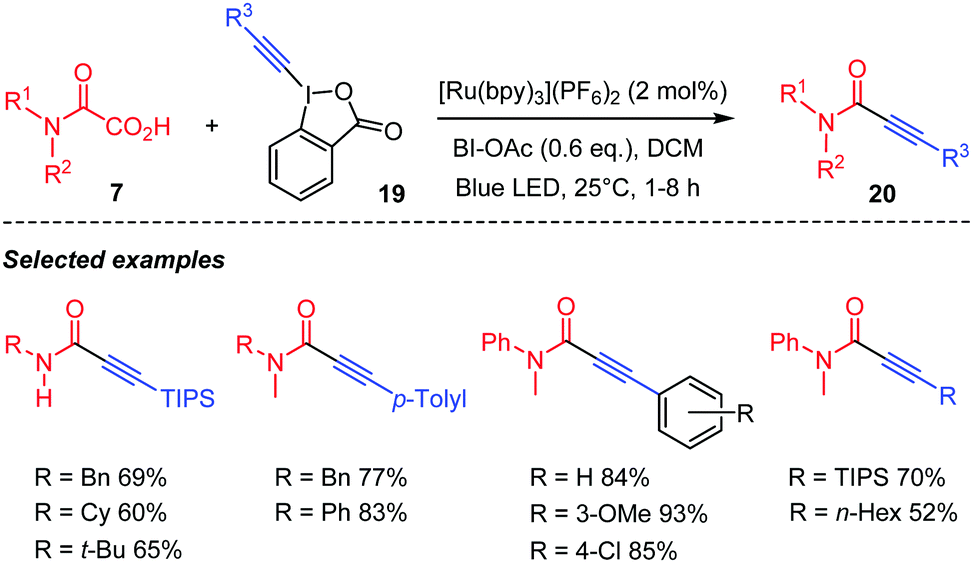 | ||
| Scheme 12 Photocatalyzed decarboxylative alkynylation of oxamic acids for the preparation of ynamides. | ||
3. Decarboxylative cyclization of oxamic acids for heterocycles synthesis
Recent years have witnessed the development of several useful synthetic procedures based on decarboxylative cyclization of oxamic acids for accessing heterocycles through C–C and C–X bonds formation. The progress in this area is summarized in this section.The first decarboxylative cyclization of oxamic acids resulting in the formation of heterocyclic compounds was reported by Minisci and co-workers in 1995.1b They showed that the Ag-catalyzed radical decarboxylation of N-methyl (or tert-butyl)-N-benzyl-oxamic acids 21a–b led to the cyclized products 22a–b alongside with homocoupling products 23a–b, though the details for the yields of the product were not available (Scheme 13). As the oxidation of the carbamoyl radical of secondary alkylamines is slow relative to that of primary ones, cyclization and dimerization of the radical was observed to afford a mixture of 22 and 23.
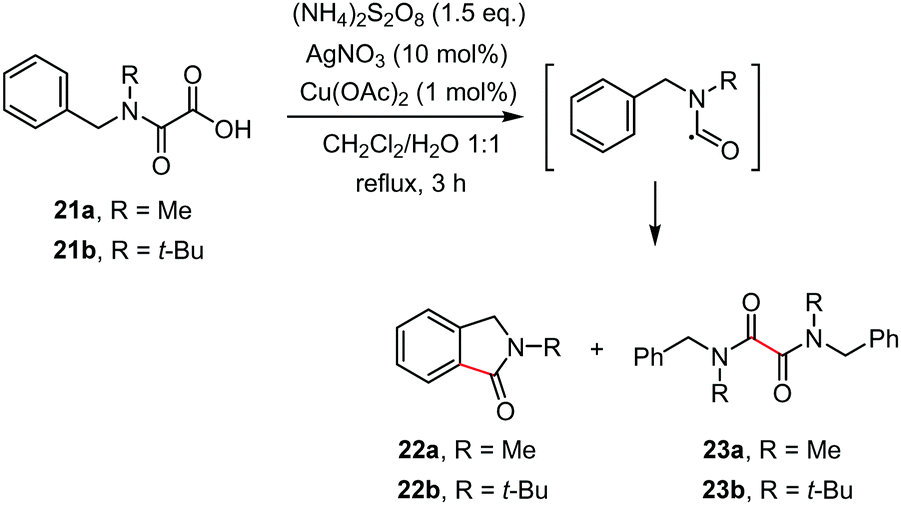 | ||
| Scheme 13 Ag/Cu-mediated decarboxylative cyclization of oxamic acids. | ||
Zhang group in 2015 developed an efficient transition-metal-free procedure for accessing phenanthridinones 25via Na2S2O8-mediated decarboxylative cyclization of biphenyl-2-oxamic acids 24 (Scheme 14).36 This simple procedure was shown to be efficient with different biphenyl-2-oxamic acids having both electron-withdrawing and electron-donating groups, furnishing the target compounds in moderate to high yields without precautions to exclude air from the reaction mixture. The authors proposed that the reaction possibly progressed through a homolytic aromatic substitution,37 involving the addition of a carbamoyl radical i onto the proximal arene, followed by the oxidation of the resulting cyclohexadienyl radical ii into cation iii, by S2O82− and rearomatization and loss of a proton.
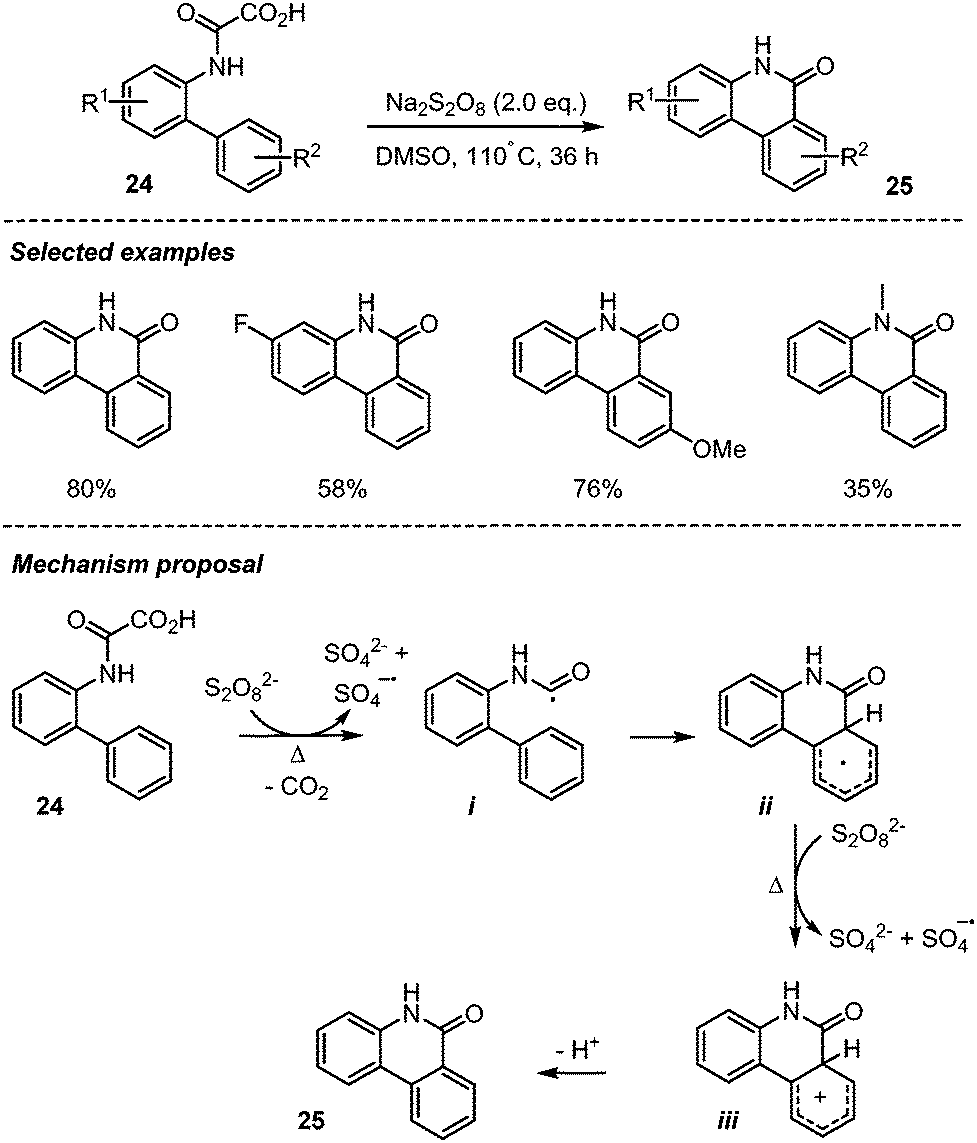 | ||
| Scheme 14 Decarboxylative cyclization of oxamic acids for phenanthridinones synthesis. | ||
The procedure was also applied for the synthesis of isoindolinone 26a as well as isoquinolinone 26b (Scheme 15).36
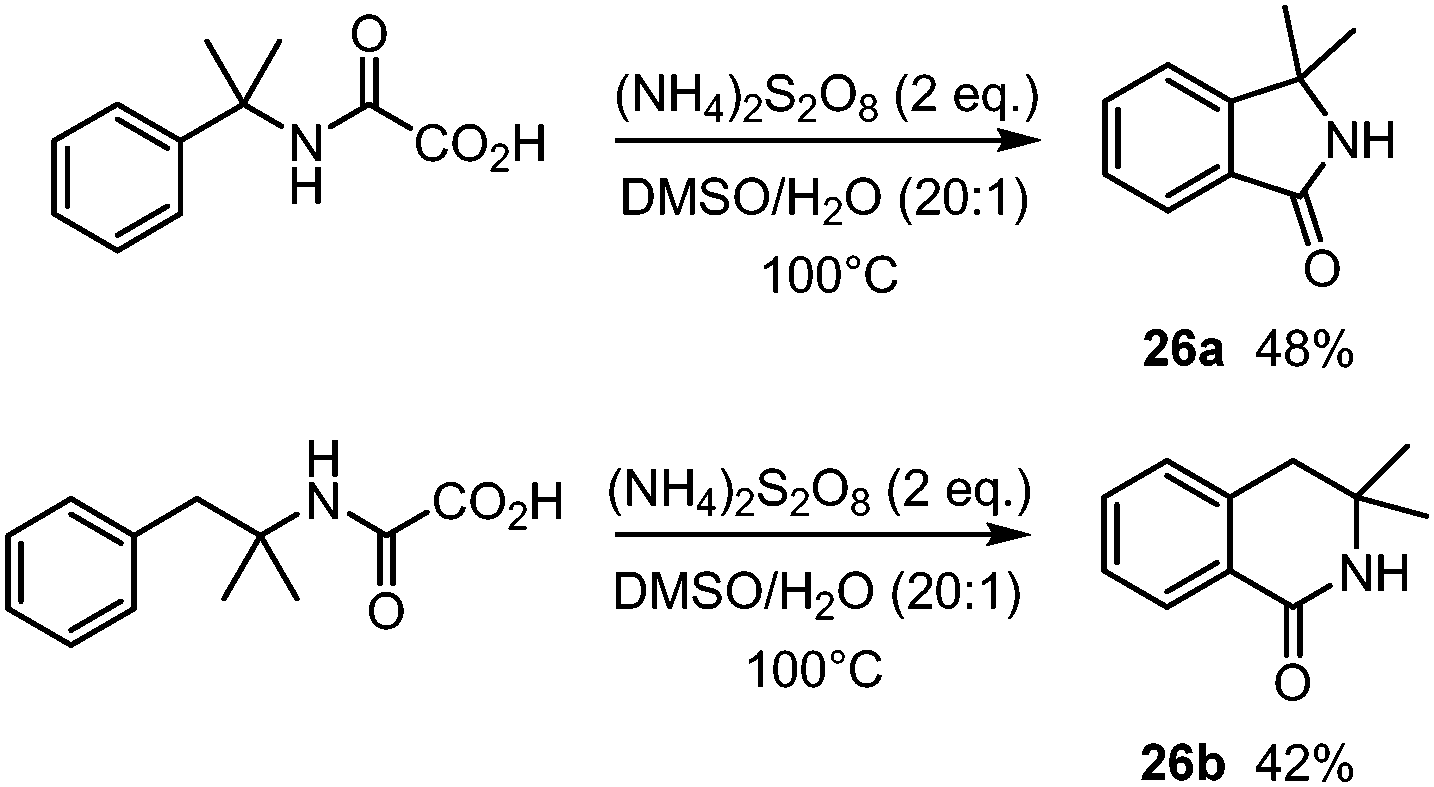 | ||
| Scheme 15 Decarboxylative cyclization of oxamic acids. An access to isoindolinones and isoquinolinones. | ||
In 2018, Wang et al., reported a hypervalent iodine(III)-mediated decarboxylative intramolecular Heck-type reaction of oxamic acids for the synthesis of 2-quinolinones, a well-known structural motif in many natural products and pharmaceutical agents (Scheme 16).38 This mild and metal-free procedure delivered a wide range of 2-quinolinones 28 in good yields from the corresponding 2-vinyl-phenyl oxamic acid 27. Usual phenyliodine(III) diacetate (PIDA) and phenyliodine(III) bis-trifluoroacetate (PIFA) reagents were found to be less efficient than the p-fluoro analogue. The authors showed that the presence of TEMPO in the reaction mixture significantly suppressed the formation of 2-quinolinones, suggesting the involvement of a radical pathway. Based on this and literature precedent, they proposed a ring-strain-enabled radical decarboxylation mechanism, in which the hypervalent iodine reagent first forms a macrocyclic iodine(III) trimer i through self-assembly. A similar macrocycle generated from amino-acids and PIDA was reported by Zhdankin et al. and its structure unambiguously assigned through X-ray diffraction studies.39 A significant high ring strain in this macrocyclic species is believed to facilitate the homolysis of the iodine-oxygen bond to generate diradical intermediate ii. This is followed by decarboxylative radical addition onto the olefin and cyclization. Subsequent intramolecular electron transfer in iii leads to a benzylic cation intermediate iv. The formation of the latter is supported by the observation that when the benzylic CH3 was replaced by –CF3, the reaction was completely shutdown. The presence of the NH group in the oxamic acid also proved very important, as the desired product was not observed when N-methyl substituted oxamic acid was used.
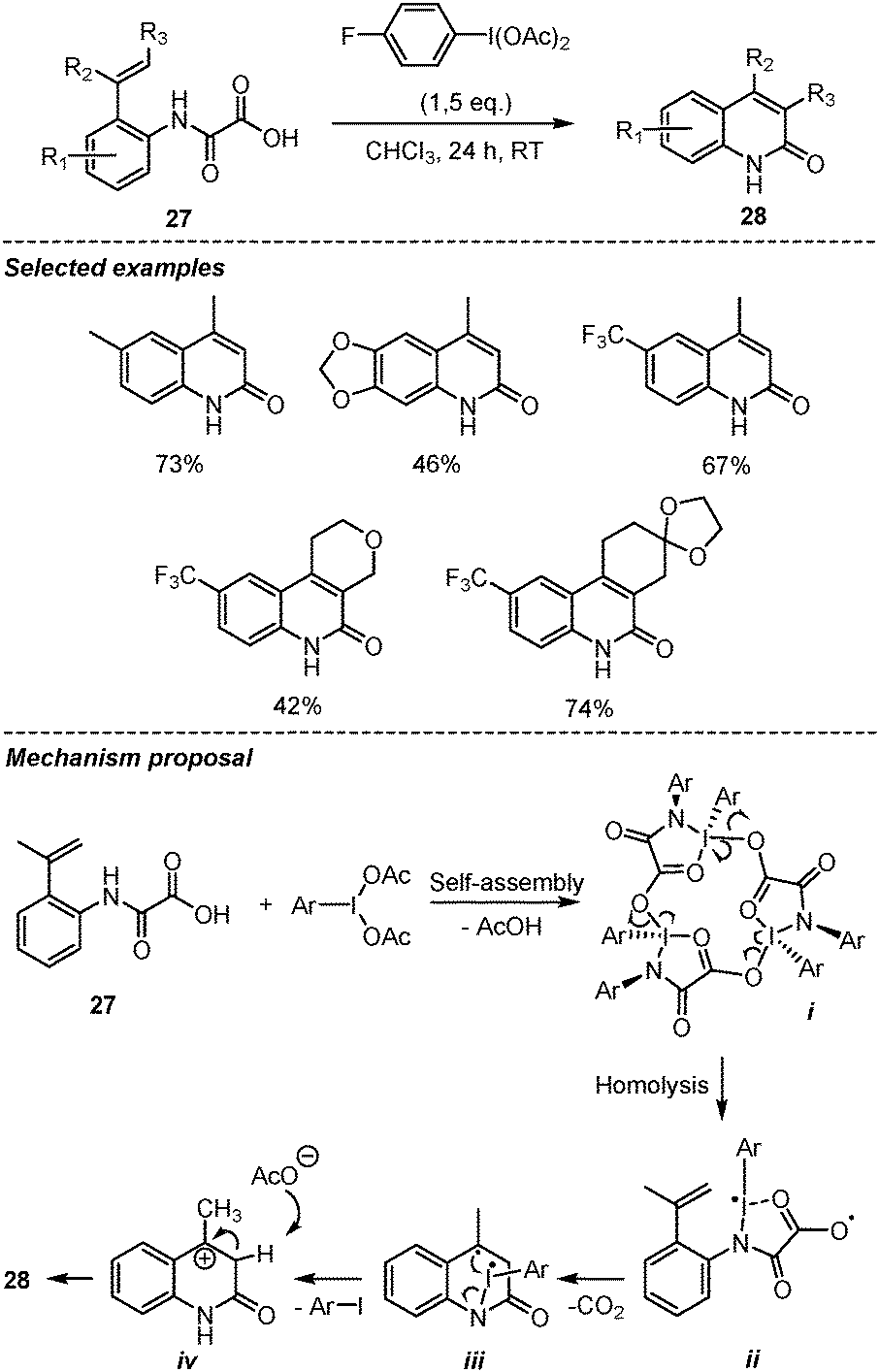 | ||
| Scheme 16 Intramolecular decarboxylative Heck-type reactions of oxamic acids. | ||
Intermolecular addition-cyclization cascade reactions using oxamic acids have also been described. These reactions are summarized below. A new and efficient procedure for the synthesis of carbamoyl quinoline-2,4-diones 31 based on persulfate mediated tandem radical cyclization of ortho-cyanoarylacrylamides 30 with oxamic acids 29 was reported (Scheme 17).40 A wide range of oxamic acids with different substituents on the phenyl ring and N-(2-cyanoaryl)acrylamides were compatible with the reaction conditions, providing the desired product in moderate to high yields. However, acrylamides with a free N–H bond were not suitable. As described above, the reaction proceeds through the (NH4)2S2O8-mediated decarboxylation of oxamic acid 29 generating the carbamoyl radical II, which adds to the ortho-cyanoarylacrylamides 30 to give an alkyl radical intermediate i. The latter then undergoes a 6-exo-dig cyclization on the nitrile group to give an iminyl radical ii, followed by H-abstraction (from the solvent) to give imine iii. The latter would finally undergo hydrolysis to deliver the final product 31.
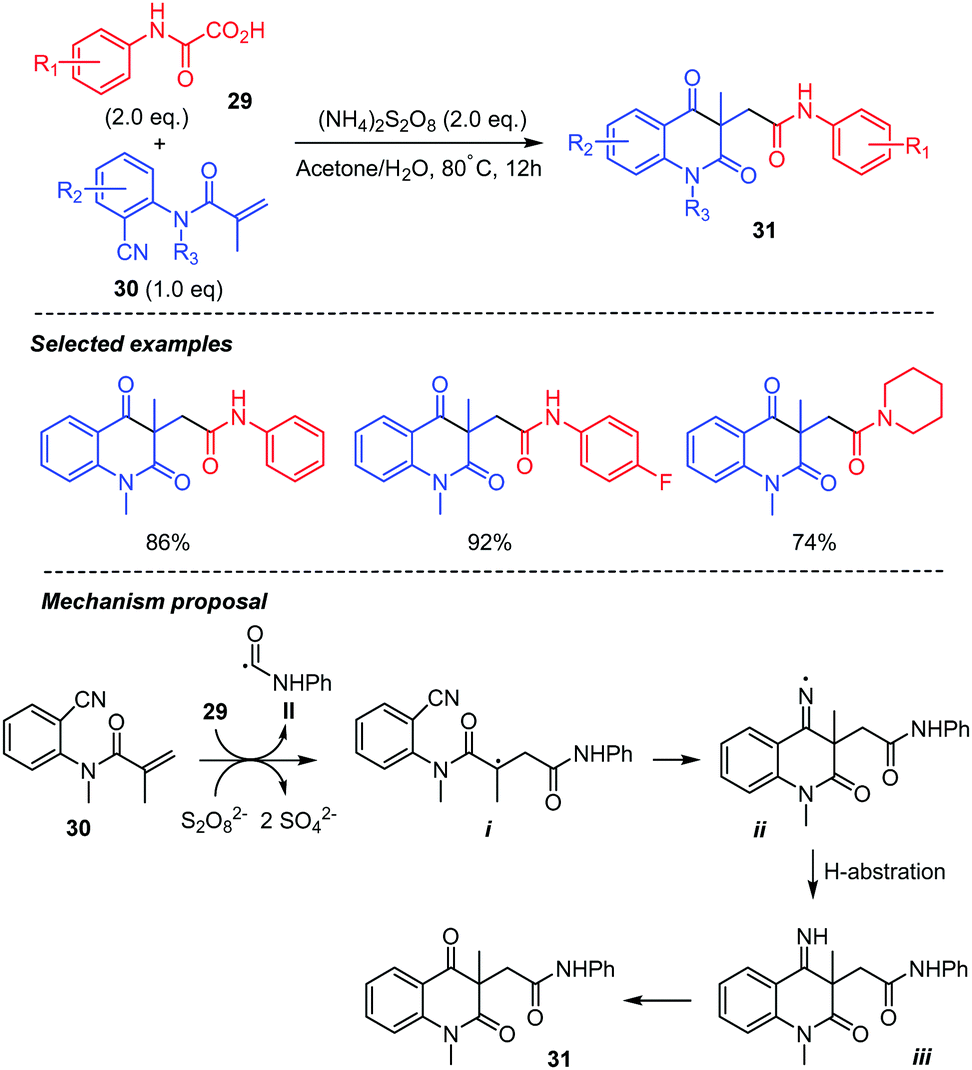 | ||
| Scheme 17 Persulfate mediated tandem addition-cyclization of carbamoyl radicals to ortho-cyanoarylacrylamides. | ||
Interestingly, when acrylamide 30 does not bear an ortho CN substituent as in 32, radical intermediate i was shown by Ma and co-workers41 to directly cyclize onto the aromatic ring providing the 5-membered ring lactam 33 in generally high yields whatever the substitution on the aromatic ring (Scheme 18). N-Protected acrylamide 32 was however compulsory as the reaction with the N–H analogue did not afford the desired lactam.
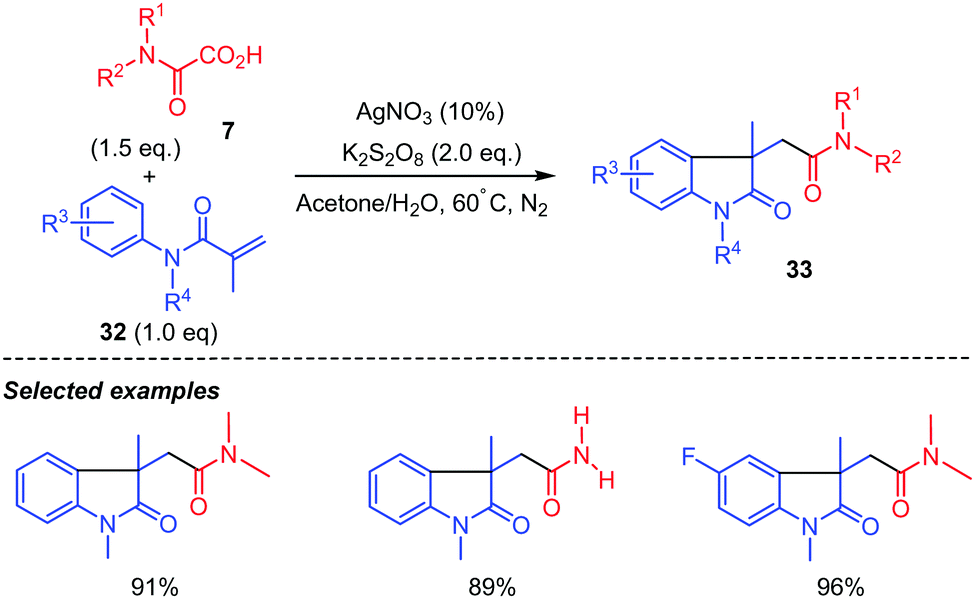 | ||
| Scheme 18 Persulfate mediated tandem addition-cyclization of carbamoyl radicals to arylacrylamides. | ||
The same authors generalized the approach to the addition of carbamoyl radical II to acrylimides 34 (Scheme 19).41 In this case, the addition was followed by a 6-exo cyclization of the radical i, onto the aromatic ring, affording the corresponding cyclohexadienyl radical ii. Oxidation of the latter led to the corresponding cation iii, giving to the 6-membered ring imide 35 after rearomatization. The scope of the reaction was shown to be rather broad, reaction conditions being compatible with electron-poor and rich substituents on the arene, the process also working with mono- and unsubstituted oxamic acids 7.
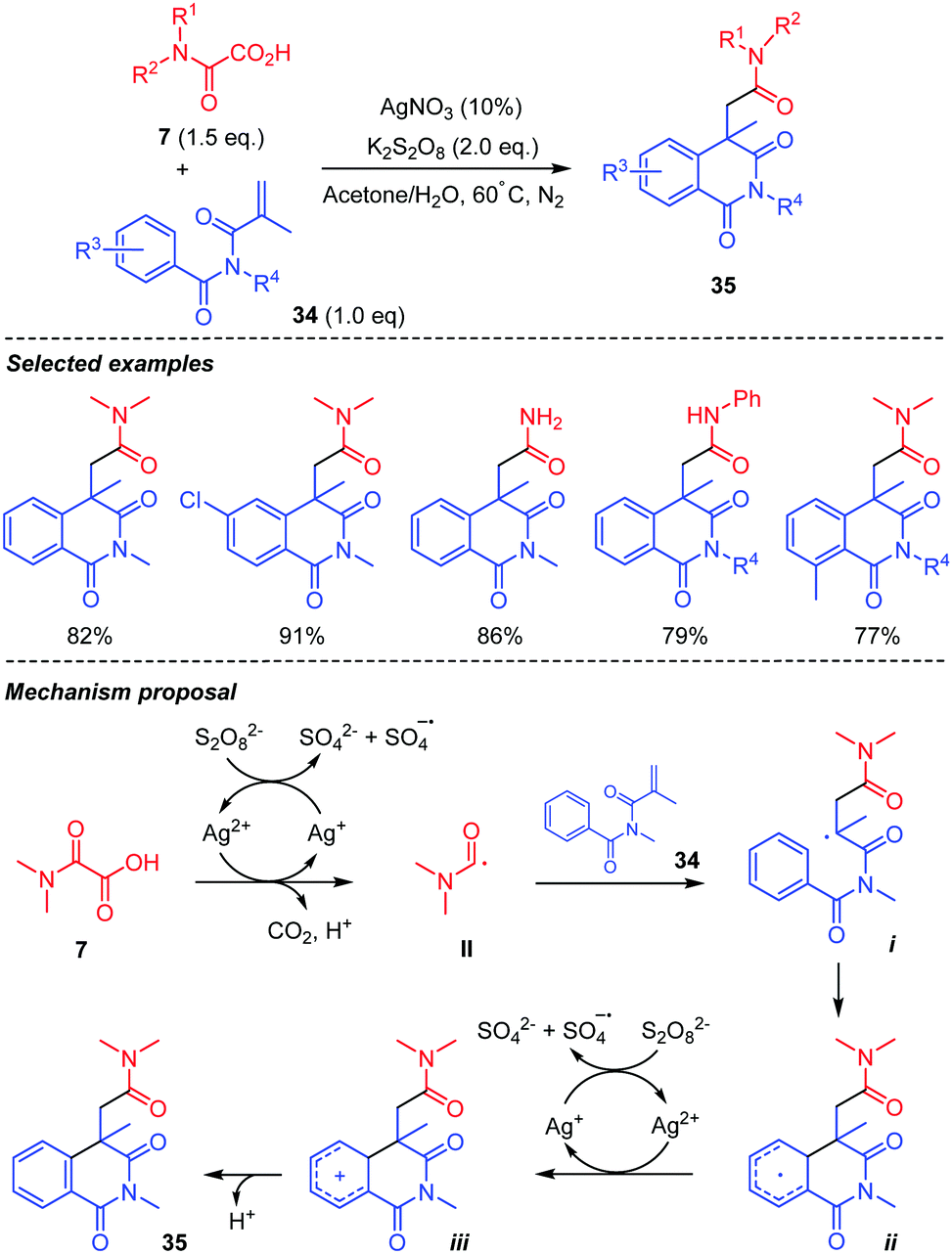 | ||
| Scheme 19 Persulfate mediated tandem addition-cyclization of carbamoyl radicals to arylacrylimides. | ||
A closely related studies was recently reported, which included the intermolecular addition of a carbamoyl radical onto an unsaturated amide attached to a benzimidazole skeleton 36 (Scheme 20).42 The carbamoyl radical II was generated as above from 7 through thermal decomposition of a persulfate in DMSO. The addition of the carbamoyl radical II onto 36 is followed by a cyclization of the resulting radical i onto the neighbouring arene to generate ii, further oxidized into the cation iii, which finally rearomatizes to afford 37.
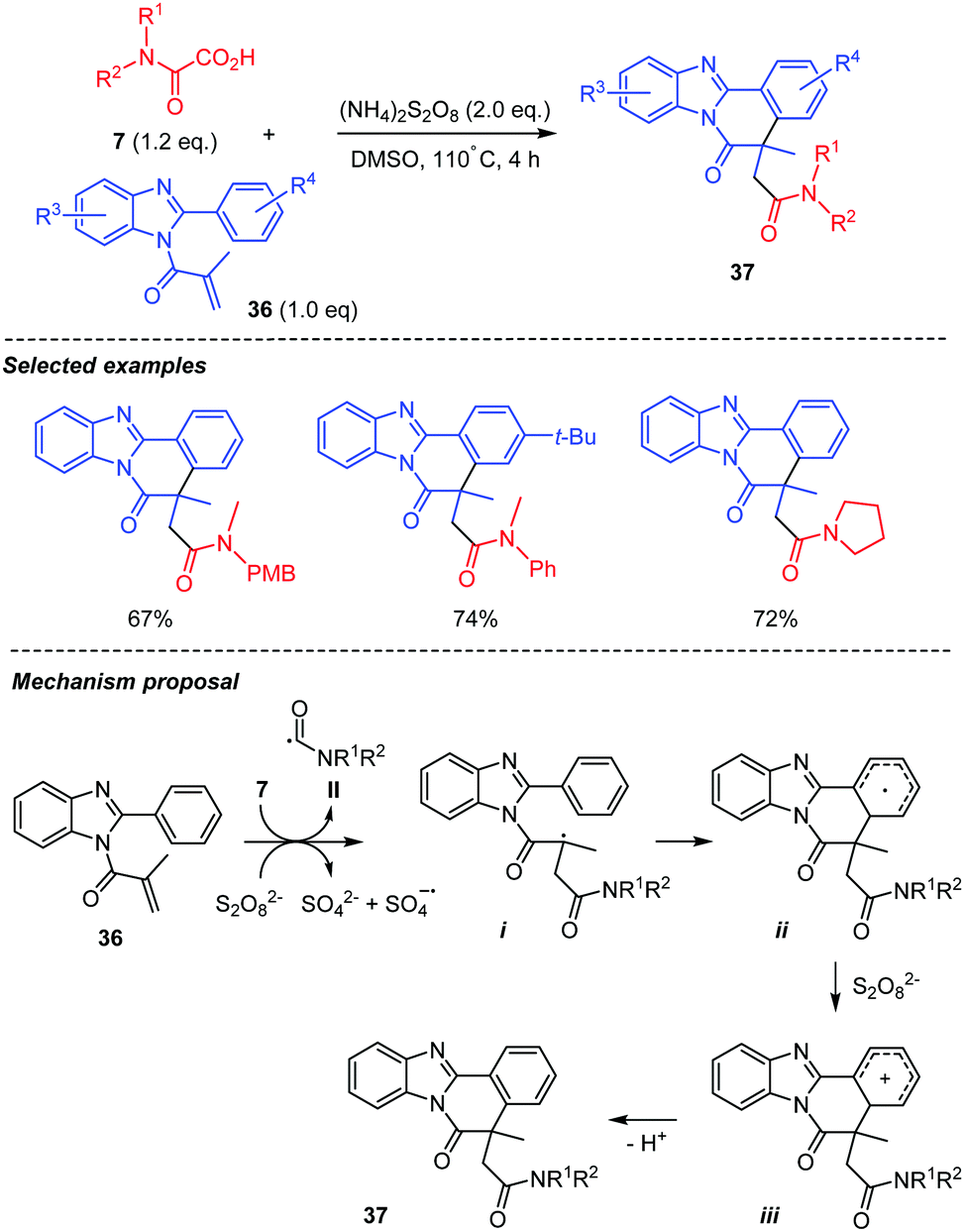 | ||
| Scheme 20 Persulfate mediated tandem addition-cyclization of carbamoyl radicals to benzimidazoylacrylamides. | ||
Interestingly, in the same study,42N-sulfonyloxamic acids 38 were submitted to the similar reaction conditions, which led to the addition of the sulfonyl group resulting from a homolytic cleavage of the N–S bond, leading to 39 (Scheme 21).
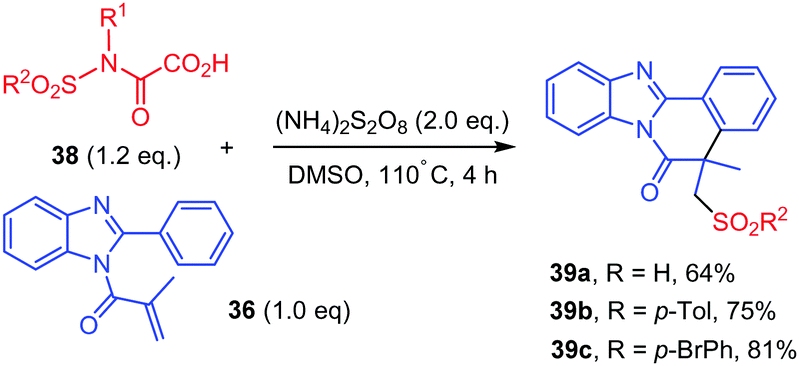 | ||
| Scheme 21 Persulfate mediated tandem addition-cyclization of sulfonyl radicals to ortho-cyanoarylacrylamides. | ||
Liu and co-workers reported a closely related Ag-promoted decarboxylative radical addition/annulation of oxamic acids 40 with gem-difluoro-olefins 41, leading to CF2-containing 3,4-dihydroquinolin-2-ones 42 (Scheme 22).43 Various N-substituted aniline-derived oxamic acids and gem-difluoroalkenes 41 bearing electron-withdrawing and electron-donating groups on the benzene ring were compatible with the reaction, furnishing the desired products in moderate to good yields. Aliphatic gem-difluoroalkenes were however not compatible with the reaction conditions. Control experiments showed that Ag(I) catalyst was essential for the reaction, as the product was not observed in its absence. Furthermore, the reaction was inhibited by radical scavengers including TEMPO and 3,5-di-tert-butyl-4-hydroxytoluene (BHT), indicating the involvement of a radical in the transformation. Based on these and pioneering work by Minisci, the authors proposed that the reaction proceeds by the generation of the carbamoyl radical-mediated by Ag(II),1 followed by an intermolecular radical addition onto the gem-difluoroalkene. This leads to a benzyl radical intermediate i, which undergoes intramolecular cyclization (to generate ii), then dehydroaromatization by the highly oxidizing SO4˙− to furnish the final product.
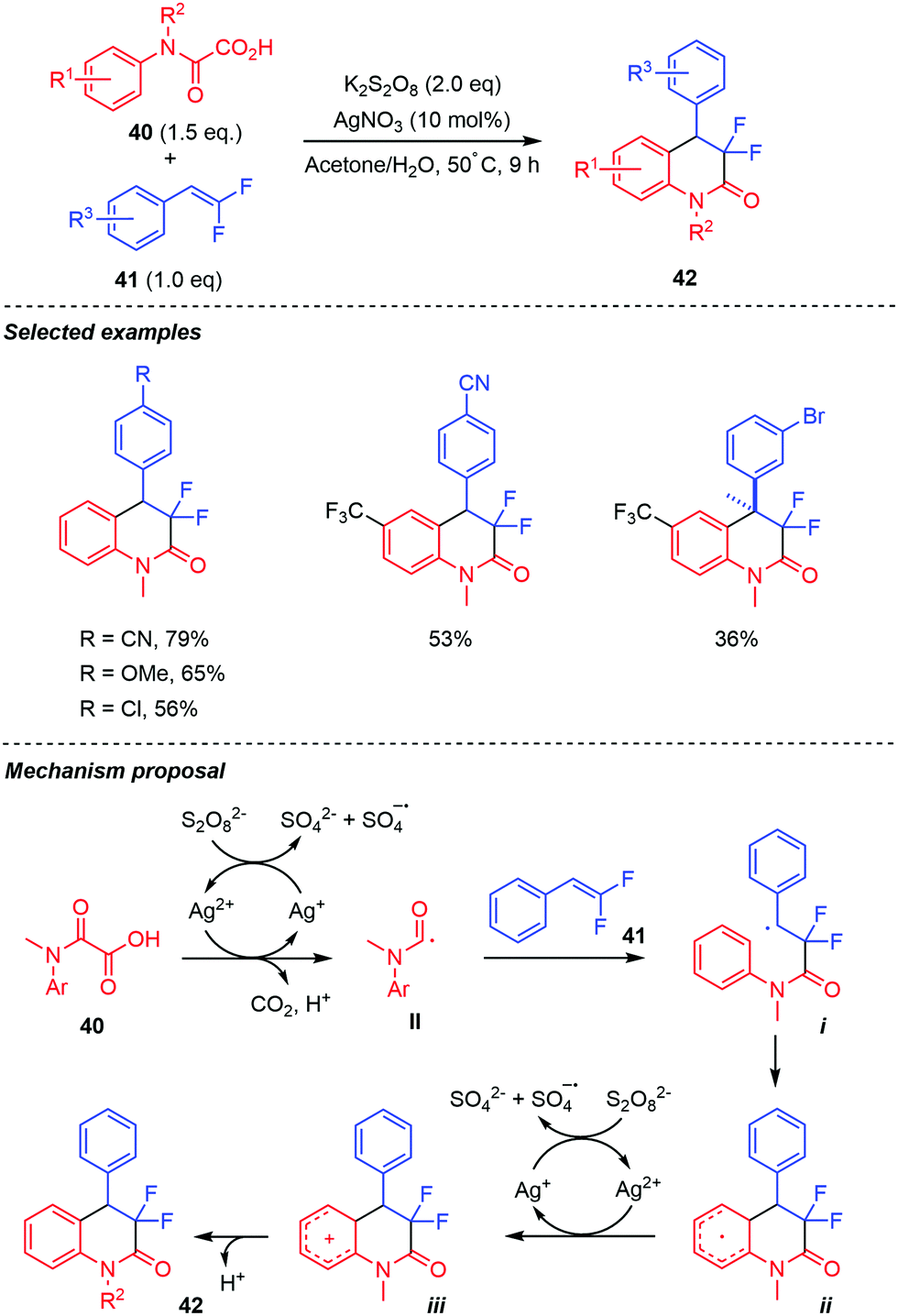 | ||
| Scheme 22 Persulfate mediated tandem addition-cyclization of carbamoyl radicals to difluorostyrenes. | ||
A similar silver-catalyzed tandem decarboxylative radical addition/cyclization of oxamic acids with simple alkenes for the synthesis of substituted 3,4-dihydroquinolin-2(1H)-ones 44 was also reported by the Feng group (Scheme 23).44 Their method demonstrated a wide substrates scope, including not only styrenes (43, R3 = Ar), but also various electron-deficient alkenes such as ethyl vinyl ketone, ethyl acrylate (43, R3 = CO2Et), and α-methylene-γ-butyrolactone, which were competent partners for the reaction, furnishing the expected dihydroquinolin-2-ones in moderate yields. In contrast, monosubstituted oxamic acids were not competent substrates for this reaction.
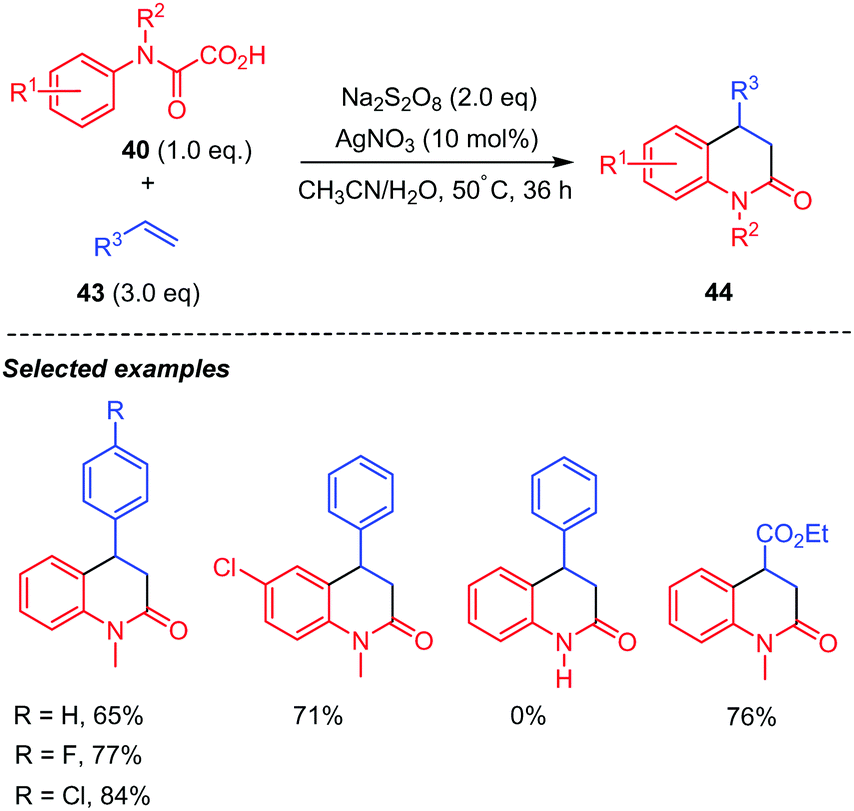 | ||
| Scheme 23 Persulfate mediated tandem addition-cyclization of carbamoyl radicals to styrenes and electron-deficient alkenes. | ||
Phenyl vinyl sulfones 45 were also good candidates for the reaction but delivered quinolin-2(1H)-ones 37 instead (Scheme 24).44 The authors proposed that the generated 4-(phenylsulfonyl)-3,4-dihydroquinolin-2(1H)-ones possibly underwent Julia–Lythgoe elimination at high temperature to give the corresponding quinolin-2(1H)-ones 46. Furthermore, control experiment indicated that AgNO3 was necessary for the reaction, as the product was not observed in its absence. In addition, methyl- and phenyl-N-protected oxamic acids displayed interesting chemoselectivity, while the benzyl counterpart gave a mixture of two products including a radical rearrangement product.
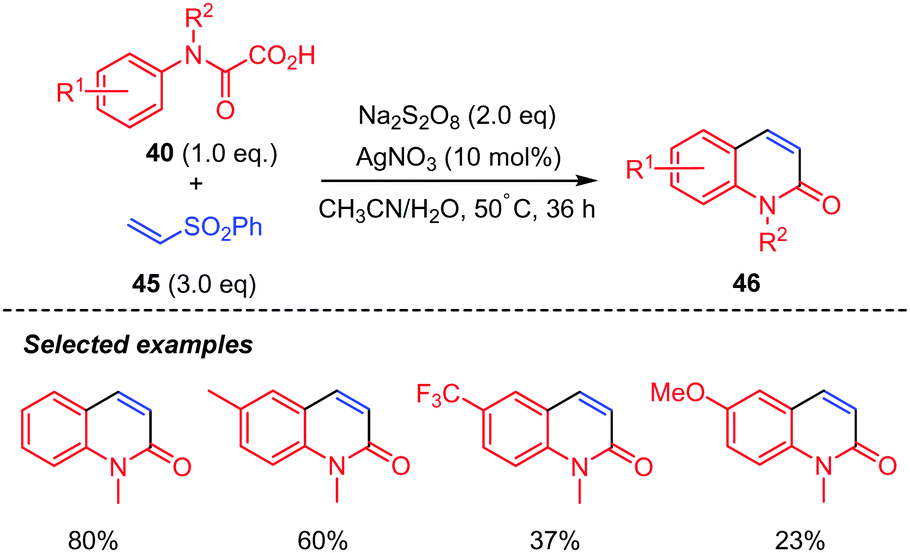 | ||
| Scheme 24 Persulfate mediated tandem addition-cyclization of carbamoyl radicals to vinylsulfones. | ||
A visible light mediated decarboxylation addition/cyclization of oxamic acids for the synthesis of 3,4-dihydroquinolin-2(1H)-ones was reported by Feng and co-workers in 2018 (Scheme 25).45 Various aniline derived oxamic acids with different substituents and electron-deficient alkenes 47, including acrylonitrile, phenyl vinyl sulfone, butyl acrylate, ethyl vinyl ketone and N,N-dimethylacrylamide, were suitable for the reaction, leading to diverse 3,4-dihydroquinolin-2(1H)-ones 48 in moderate yields. The authors proposed that the transformation proceeds by oxamic acid decarboxylation mediated by the Ir(III)* catalyst in its excited state. The carbamoyl radical II thus generated would undergo intermolecular addition to the electron deficient alkene 47 to give radical intermediate i, which would cyclize leading to the cyclohexadienyl radical ii. Oxygen present in the medium would play the role of an external oxidant by regenerating the reduced catalyst Ir(II) in its ground state Ir(III). The oxygen radical anion generated would then abstract a proton from ii to afford the product 48 and H2O2. Interestingly, the photochemical conditions described here provide a different outcome for the reaction involving a vinylsulfone such as 45 (Scheme 24), leading to the 4-(phenylsulfonyl)-3,4-dihydroquinolin-2(1H)-ones instead of the elimination as mentioned above using persulfate conditions.
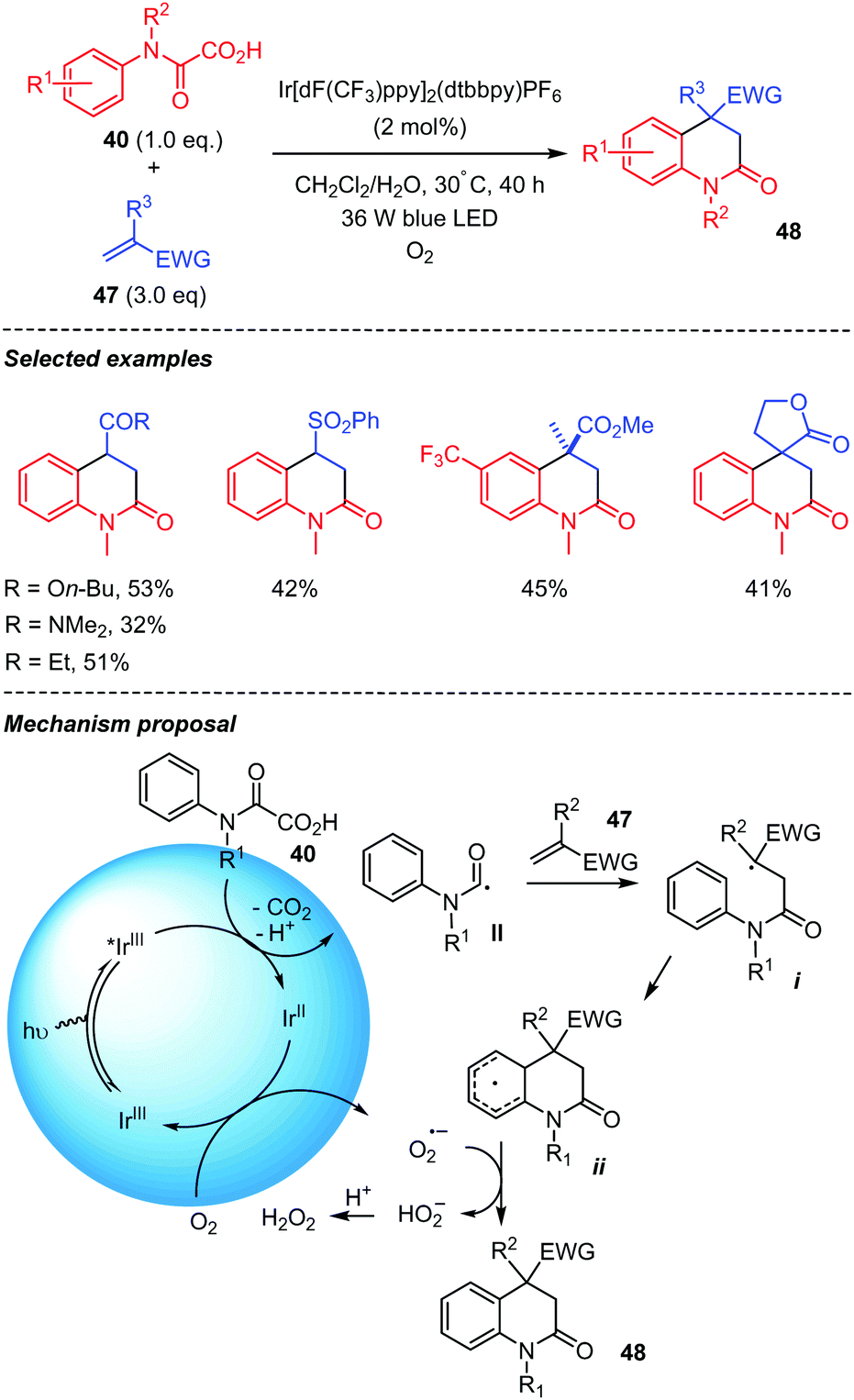 | ||
| Scheme 25 Visible-light-mediated tandem addition-cyclization of carbamoyl radicals onto electron-deficient alkenes. | ||
Pedersen et al. recently reported on a double decarboxylative addition of oxamic acids onto acrylic acids (Scheme 26).46 This addition-cyclization cascade provided a series of quinoline-2-ones in good yields. The process is reminiscent to the one described in Scheme 24, where a sulfone moiety was eliminated in the last stage of the process. The reaction may be performed under thermal or photocatalytic conditions as summarized in Scheme 26. The organophotocatalyst (4-CzIPN) in its excited state (PC*) reduces the persulfate into the sulfate radical anion SO4˙−, which then oxidizes Ag(I) in to Ag(II). The latter generates the carbamoyl radical II from oxamic acid 40, which adds to the acrylic acid 49. The addition-cyclization process then affords intermediate i, the oxidation of which with PC+˙ and loss of a proton give dihydroquinolin-2-one ii, returning the photocatalyst in its ground state. A similar decarboxylation-elimination pathway finally converts ii into 50 through iii. Under thermal conditions, the authors propose that H-abstraction by SO4˙−, issued from thermolysis of S2O82−, replaces SET and loss of proton.
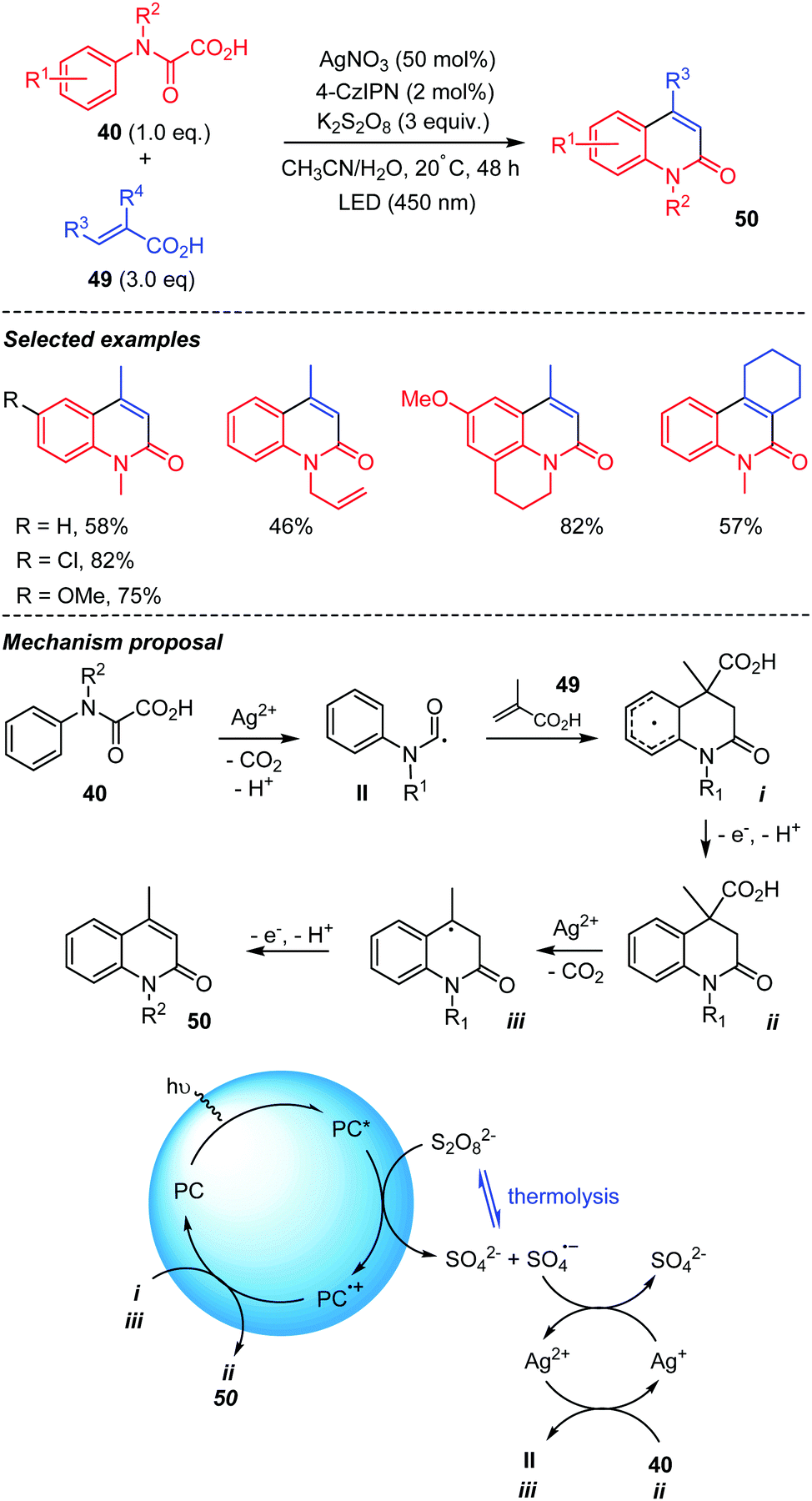 | ||
| Scheme 26 Photocatalyzed-Persulfate mediated tandem addition-cyclization of carbamoyl radicals to acrylic acids. | ||
4. Oxamic acids as precursors of isocyanates, urethanes, and ureas
Decarboxylation of oxamic acids and further oxidation of the carbamoyl radical were originally shown by Minisci et al. to afford a straightforward entry towards isocyanates.1 Therefore, oxamic acids have recently been designated as potent green precursors for the synthesis of urethanes and ureas through in situ generation of isocyanates using a non-phosgene route.21 This strategy is particularly relevant as it avoids the manipulation of carcinogenic isocyanates and highly toxic gaseous phosgene. Minisci and co-workers originally showed that oxamic acids can be oxidized into isocyanates using S2O82− as an oxidant, AgNO3 and Cu(OAc)2 as catalysts, and a biphasic medium involving water and an organic solvent (Scheme 27).1 Various isocyanates 52 could be accessed with their method in moderate to good yields using different monooxamic acids 51. Interestingly, oxidation of secondary oxamic acid into isocyanate was not feasible, pointing toward the importance of the –NHCO– group in the second oxidation process. The silver salt was vital for the reaction as no isocyanates were observed in its absence, while Cu(OAc)2 was required to favor the slow oxidation of the carbamoyl radical II into isocyanate, thereby minimizing the homocoupling and the formation of the corresponding diamide (i.e.23a–b, Scheme 13).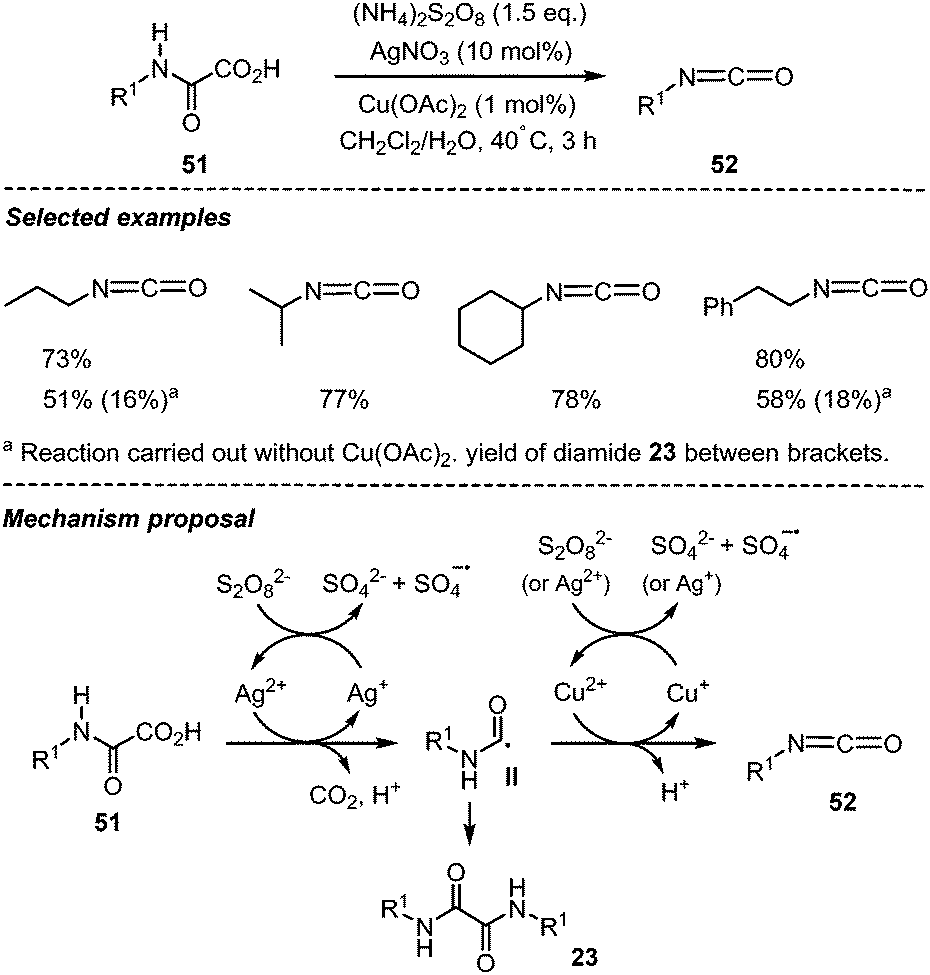 | ||
| Scheme 27 Persulfate mediated generation of isocyanate from oxamic acids. | ||
Although this method gave access to isocyanates, yields were generally moderate due to partial hydrolysis of the generated isocyanate into amine in the aqueous medium. Moreover, direct access to urethane using this method was not feasible, leaving the option of isolating and purifying the generated carcinogenic isocyanate.
Recently our group developed a metal-free photocatalyzed procedure for direct access to urethanes and ureas from oxamic acids (Scheme 28).21 This mild and efficient method uses an organic dye as photocatalyst, hypervalent iodine reagent as oxidant and visible-light irradiation to trigger the free-radical oxidation of oxamic acids 51 into in situ generated isocyanates in an organic solvent. In the presence of alcohols 53, the reaction furnished urethanes 54 in a one-pot process, thereby avoiding the isolation and purification of the generated isocyanate. 4-CzIPN was shown to be the most efficient organic photocatalyst for the reaction though Ru(bpy)3Cl2 and AcrMes+ClO4− could also serve as competent photocatalysts for the reaction. On the other hand, Eosin-Y and rose-Bengal gave only trace amount of the desired product. Furthermore, BI-OAc demonstrated superior performance as an oxidant for the reaction compared to other hypervalent iodine reagents tested including hydroxybenziodoxolone (BI-OH), PIDA and PIFA. The reaction worked efficiently with DCE or DCM as solvent while THF, CH3CN or DMF resulted in low product yield and DMSO failed to provide the product. This new photocatalyzed procedure exhibited a wide substrate scope. A broad variety of oxamic acids and alcohols were compatible with the system furnishing the desired urethanes in good to excellent yields with high functional group tolerance. Based on experimental evidences, a mechanism was proposed suggesting that the photoexcited catalyst (PC*) was quenched by intermediate i, formed through ligand exchange between oxamic acid 51 and BI-OAc. This led to an unstable radical-anion ii, which collapsed into carbamoyl radical II and o-iodobenzoic acid anion (Scheme 28). Further oxidation of II by the photocatalyst radical cation PC˙+ then afforded the corresponding protonated isocyanate iii, while the photocatalyst was regenerated. The protonated isocyanate can either reacts in situ with the alcohol 53 to furnish the desired urethane 54 or loses a proton to give isocyanate 52, which is effectively isolated in good yield when the reaction is performed in the absence of alcohols, further supporting the mechanism below.
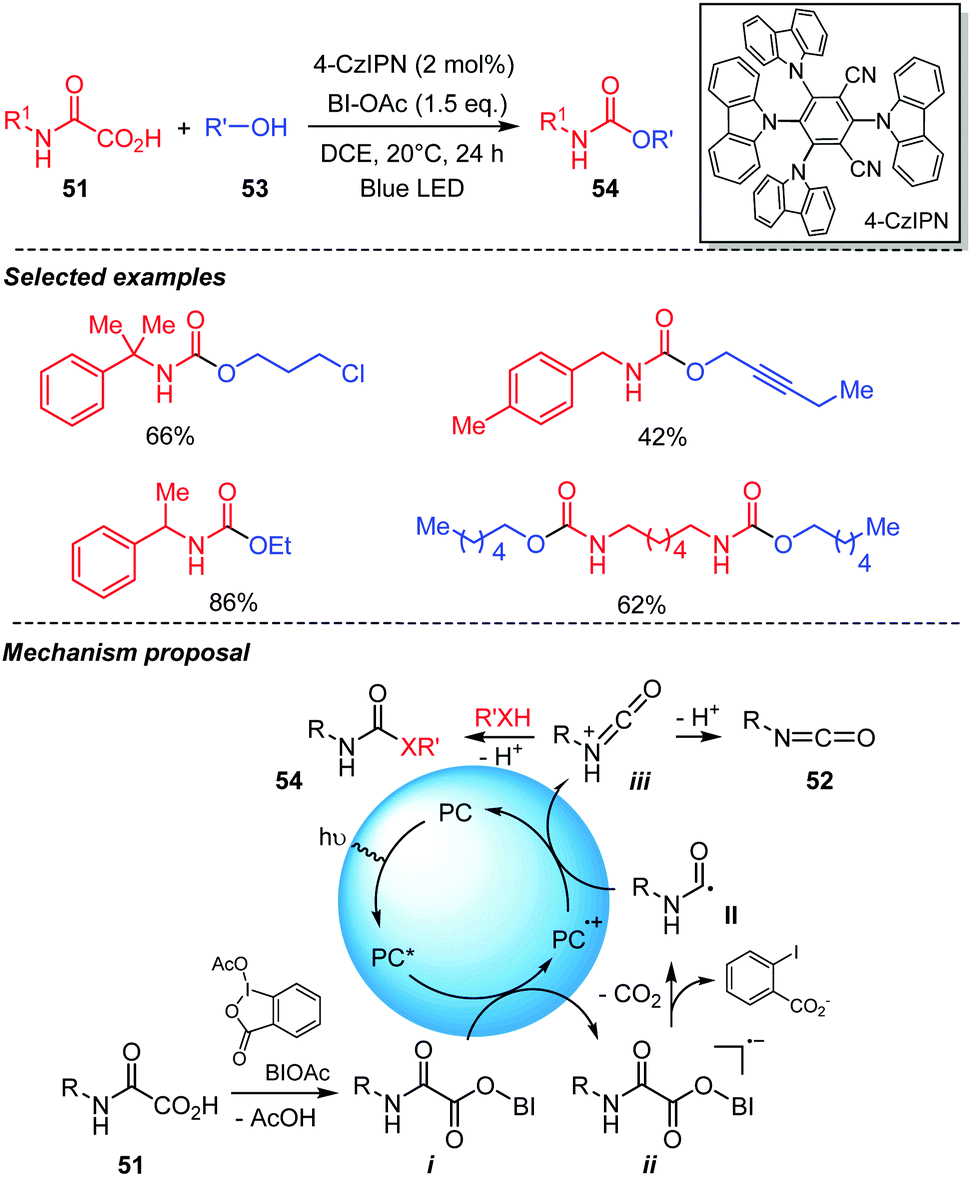 | ||
| Scheme 28 Urethane synthesis through visible-light mediated decarboxylation of oxamic acids. | ||
Extension of the process to the preparation of unsymmetrical ureas 56 was also performed successfully (Scheme 29). However, in this case, a two-step one pot protocol was adopted, in which isocyanate 52 was first generated in situ under photocatalytic conditions, and then the amine 55 added to furnish the corresponding urea 56 without isolation and purification of the generated isocyanate. A recent report by Li and co-workers described a similar work using BI-OAc as an oxidant and a (polyaniline)-g-C3N4–TiO2 composite as a photocatalyst.47 Addition of the amine as above, after the photocatalytic process led to various unsymmetrical ureas in satisfying yields.
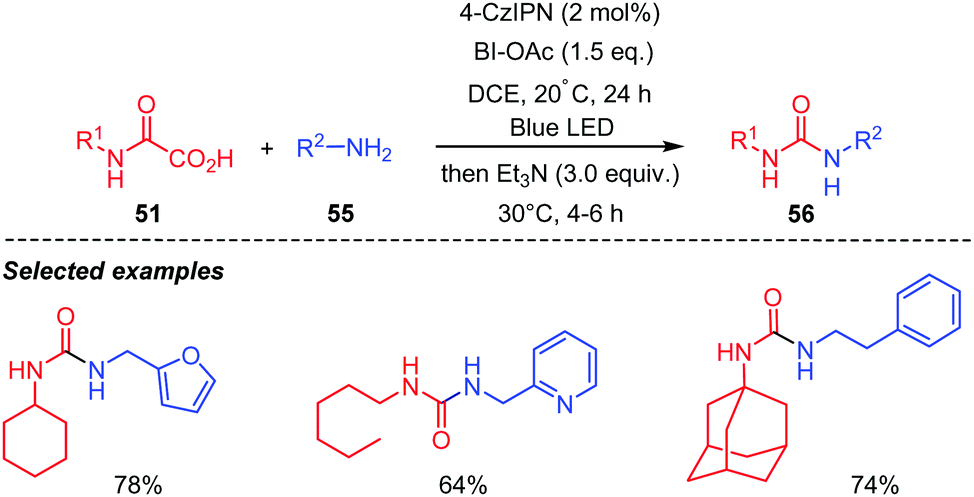 | ||
| Scheme 29 Urea synthesis through visible-light mediated decarboxylation of oxamic acids. | ||
Pursuing in this direction, our laboratory also developed an electrochemical version of the above reaction, in order to replace the hypervalent iodine oxidant by an anodic oxidation. This metal-, photocatalyst-, light and chemical oxidant-free procedure thus led to a broad range of urethanes, including some prepared from chiral oxamic acids (Scheme 30).48 The latter were accessible without racemization. Simple inexpensive graphite anodes and cathodes in an undivided cell were used in this context. Alcohols were generally present as solvents, but the excess could be recovered by simple distillation at the end of the process. With MeOH as a solvent the reaction did not require any electrolyte, while n-Bu4NCl and a gentle heating were necessary with other less conducting alcohols. This Hofer–Moest process proceeds through the oxidation at the anode of the oxamic acid into the carboxyl radical i, which upon decarboxylation provides the carbamoyl radical II. Further oxidation of the latter at the anode finally generates the cationic species ii, which reacts with alcohols affording the desired urethanes. Evidences for this mechanism was supported by the isolation of a bis-amide (such as 23a–b, Scheme 13) resulting from the dimerization of the carbamoyl radical II, when the reaction was carried out with a minimum amount of alcohol. The occurrence of cation iii was supported by the isolation of a diethylurea resulting from the reaction of iii with diethylamine issued for the anodic oxidation of NEt3 used as a base. As observed by Minisci, the carbamoyl radical oxidation is slow, explaining the formation of the bis-amide in some cases.
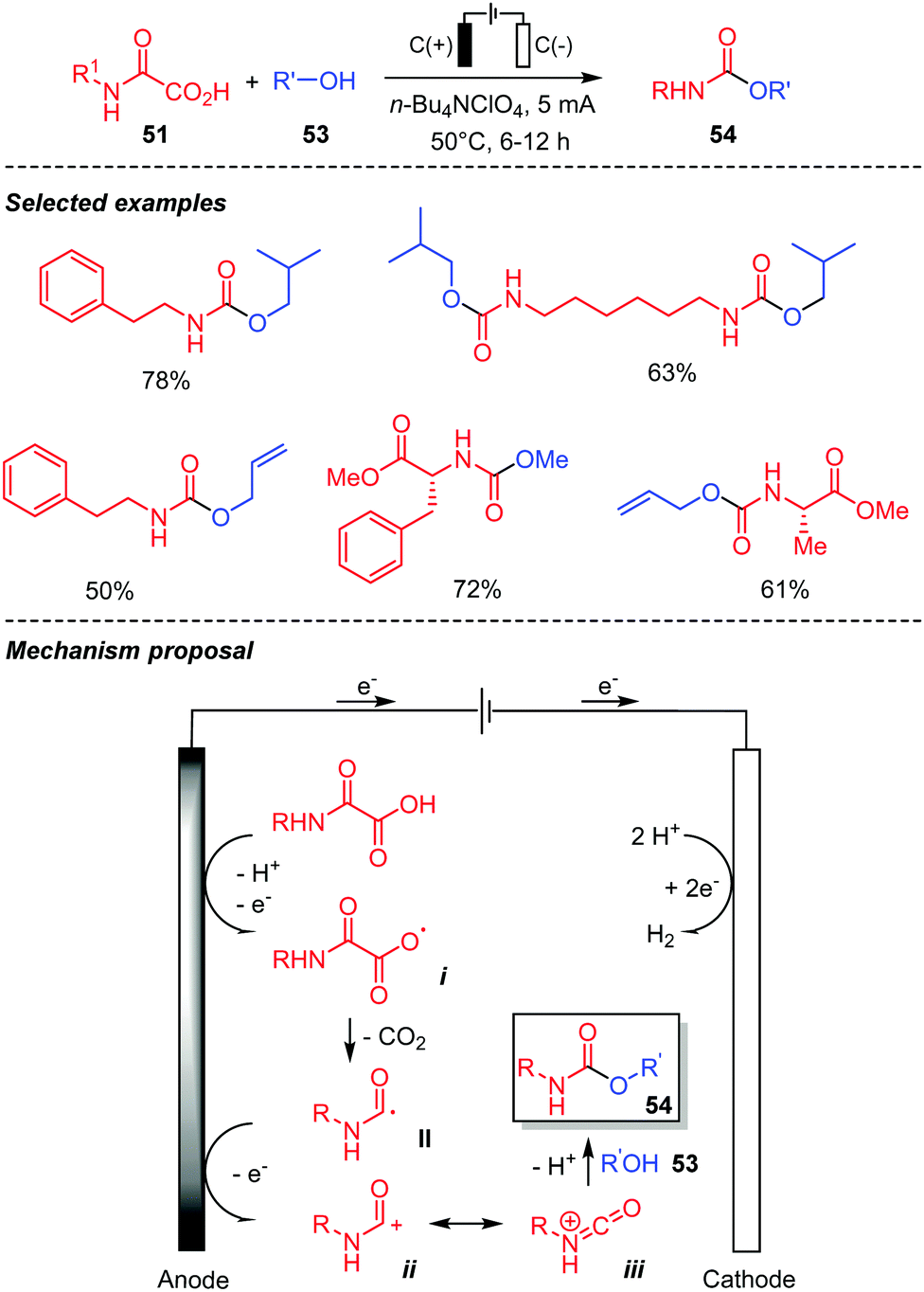 | ||
| Scheme 30 Electrochemical decarboxylation of oxamic acids in the presence of alcohols. | ||
Lam and co-workers49 recently reported a modified version of this electrochemical process, by adding collidine as an anodically stable base. The reaction led to the isocyanate, which was directly trapped by a nucleophile after the electrolysis. Various amines 55 were thus added leading to the corresponding ureas 56 in high yields (Scheme 31). The process was compatible with protected amines and also afforded unsaturated ureas, albeit with lower yields. When the nucleophile is an alcohol 53, the addition onto the isocyanate required heating and the presence of a tin catalyst (DBTDL: dibutyltin dilaurate). Chiral oxamic acids, free hydroxy groups, and alkyne substituents were compatible with the reaction conditions. The reaction was also extended to the preparation of thiocarbamates 58 by the addition of thiols 57. Finally, the authors were able to translate successfully their process to continuous flow technology.
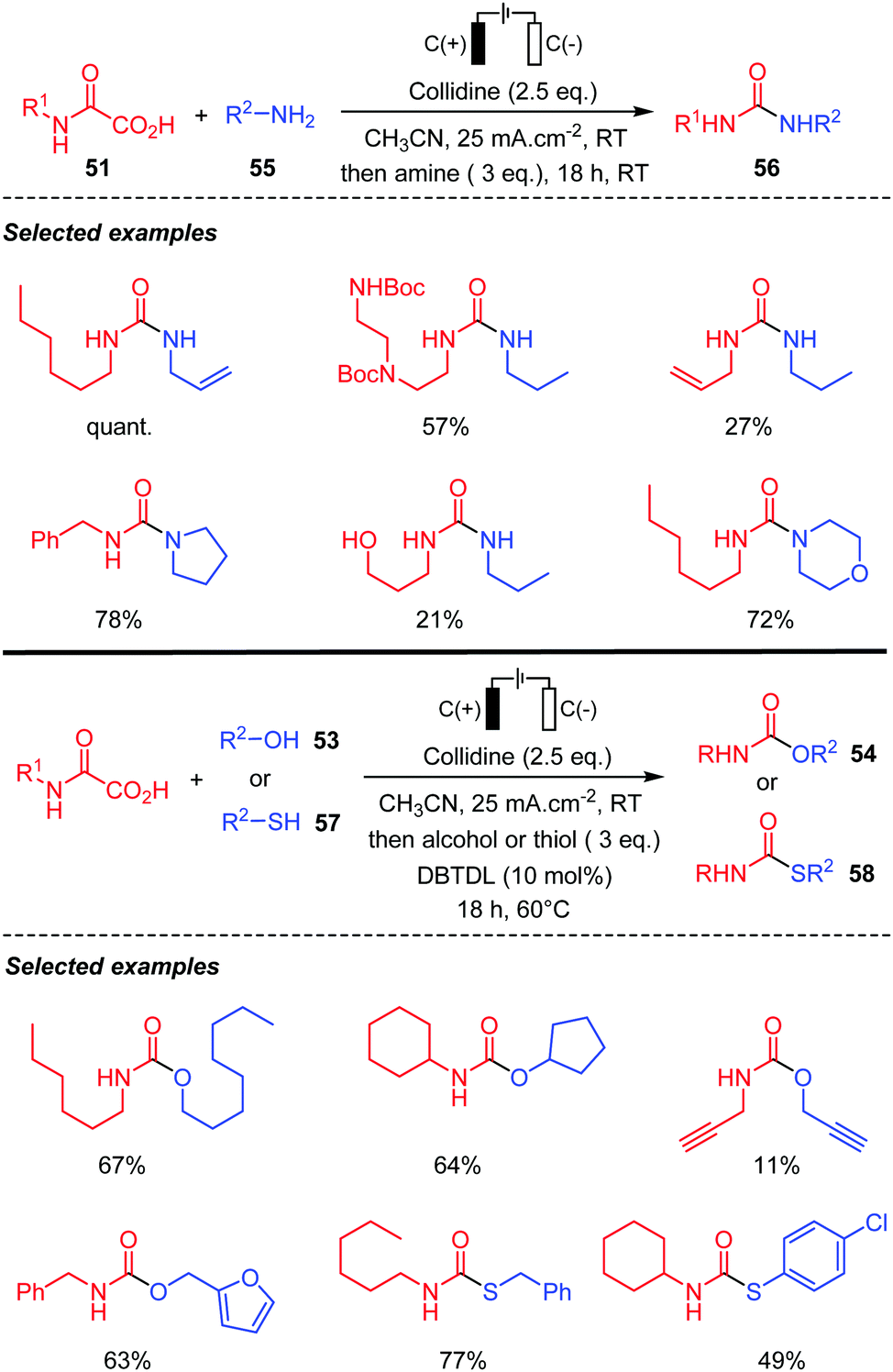 | ||
| Scheme 31 Electrochemical decarboxylation of oxamic acids in the presence of amines, alcohols and thiols using collidine as a base. | ||
5. Conclusions
In summary, oxamic acids chemistry has recently experienced a renaissance after the pioneering work of Minisci on silver-catalyzed decarboxylation.1 These readily available amido-acids proved to be environmentally benign precursors of carbamoyl radicals, capable of replacing most of the precursors previously used, which often required several synthetic steps. The few examples described above demonstrate that these nucleophilic radicals may be generated under mild conditions, in the presence of an oxidant. The latter is generally a persulfate, although hypervalent iodine reagents have demonstrated their usefulness in this context. Anodic oxidation also allows the presence of an oxidant in a stoichiometric quantity to be dispensed with. The activation of oxamic acid in the presence of the oxidant can either be thermal or photochemical. It is worth noticing that the use of silver salts to decompose persulfate is now unnecessary and has been superseded by the simple heating of the persulfate with DMSO.24–26 The radicals thus produced add efficiently to a broad range of unsaturated systems, including alkenes, both electron-rich and electron-poor and aromatic heterocycles (Minisci reaction) giving access to diversely functionalized amides. Finally, oxidation of oxamic acids in the presence of alcohols, amines, or thiols gives access to the corresponding urethanes, ureas and thioureas.21,47–49 With these new conditions in hand, the chemistry of oxamic acids and carbamoyl radicals should find new directions and original applications in the not too distant a future, including addition reactions to various unsaturated systems not investigated until now. The use of the CO2, released during the decarboxylation process, as a C1 synthon in a radical process would be worth looking at, offering an access to new structures limiting the carbon waste. Finally, metal-catalyzed coupling involving carbamoyl radicals has been overlooked so far and would certainly open new directions in the synthesis of functionalized amides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
IMO and GK respectively thank the Alex-Ekwueme Federal University Ndufu-Alike Ikwo (AE-FUNAI) and the TUBITAK BIDEB-2219 (grant no. 1059B191900553) for PhD and post-doctoral funding. We gratefully acknowledge the ANR (NCO-INNOV, No. 20-CE07-0015-01), the University of Bordeaux and the CNRS for financial support.Notes and references
- (a) F. Minisci, F. Coppa and F. Fontana, J. Chem. Soc., Chem. Commun., 1994, 679 RSC; (b) F. Minisci, F. Fontana, F. Coppa and Y. M. Yan, J. Org. Chem., 1995, 60, 5430–5433 CrossRef CAS.
- (a) D. Elad and J. Rokach, J. Org. Chem., 1964, 29, 1855–1859 CrossRef CAS; (b) D. Elad and J. Rokach, J. Org. Chem., 1965, 30, 3361–3364 CrossRef CAS; (c) D. Elad and J. Rokach, J. Chem. Soc., 1965, 800–802 CAS.
- (a) L. Friedman and H. Shechter, Tetrahedron Lett., 1961, 2, 238–242 CrossRef; (b) R. Dowbenko, Tetrahedron, 1964, 20, 1843–1858 CrossRef CAS; (c) J. Rokach and D. Elad, J. Org. Chem., 1966, 31, 4210–4215 CrossRef CAS; (d) W.-P. Mai, G.-C. Sun, J.-T. Wang, G. Song, P. Mao, L.-R. Yang, J.-W. Yuan, Y.-M. Xiao and L.-B. Qu, J. Org. Chem., 2014, 79, 8094–8102 CrossRef CAS; (e) Q. Jiang, J. Jia, B. Xu, A. Zhao and C.-C. Guo, J. Org. Chem., 2015, 80, 3586–3596 CrossRef CAS; (f) H. Wang, L.-N. Guo, S. Wang and X.-H. Duan, Org. Lett., 2001, 17, 3054–3057 CrossRef PubMed; (g) J. C. Walton, Acc. Chem. Res., 2014, 47, 1406–1416 CrossRef CAS PubMed.
- (a) T. Yonezawa, I. Nodaand and T. Kawamura, Bull. Chem. Soc. Jpn., 1969, 42, 650–657 CrossRef CAS; (b) S. R. Bosco, A. Cirillo and R. B. Timmons, J. Am. Chem. Soc., 1969, 91, 3140–3143 CrossRef CAS; (c) H. Hefter and H. Fischer, Chem. Ber., 1970, 74, 493–500 CAS; (d) G. A. DiLabio, E. M. Scanlan and J. C. Walton, Org. Lett., 2005, 7, 155–158 CrossRef CAS PubMed.
- L. Grossi, J. Chem. Soc., Chem. Commun., 1989, 1248–1250 RSC.
- R. Sutcliffe and K. U. Ingold, J. Am. Chem. Soc., 1981, 103, 7686–7687 CrossRef CAS.
- (a) G. Bryon Gill, G. Pattenden and S. J. Reynolds, J. Chem. Soc., Perkin Trans. 1, 1994, 369–378 RSC; (b) S. Guo, X. Wang, D. Zhao, Z. Zhang, G. Zhang, S. Tang and K. Sun, Asian J. Org. Chem., 2022, 11, e202100812 CAS.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2070 CrossRef CAS PubMed.
- (a) F. Minisci, A. Citterio, E. Vismara and C. Giordano, Tetrahedron, 1985, 41, 4157–4170 CrossRef CAS; (b) F. Minisci, F. Recupero, C. Punta, C. Gambarotti, F. Antonietti, F. Fontana and G. F. Pedulli, Chem. Commun., 2002, 2496–2497 RSC; (c) J. Joseph and A. P. Antonchick, Top. Heterocycl. Chem., 2018, 54, 93–149 CrossRef CAS; (d) T. Caronna, C. Gambarotti, L. Palmisano, C. Punta and F. Recupero, Chem. Commun., 2003, 2350–2351 RSC; (e) I. Kim, G. Kang, K. Lee, B. Park, D. Kang, H. Jung, Y.-T. He, M.-H. Baik and S. Hong, J. Am. Chem. Soc., 2019, 141, 9239–9248 CrossRef; (f) J.-K. Cheng, L. Shen, L.-H. Wu, X.-H. Hu and T.-P. Loh, Chem. Commun., 2017, 53, 12830–12833 RSC; (g) J. Bergès, Y. Zaïd, A. Tlili, J.-M. Sotiropoulos and M. Taillefer, Eur. J. Org. Chem., 2021, 1559–1563 CrossRef; (h) X.-Z. Fan, J.-W. Rong, H.-L. Wu, Q. Zhou, H.-P. Deng, J. D. Tan, C.-W. Xue, L.-Z. Wu, H.-R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS PubMed; (i) Y. Zhang, S. Zhang, G. Xu, M. Li, C. Tang and W. Fan, Org. Biomol. Chem., 2019, 17, 309–314 RSC; (j) Y. Zhang, K. B. Teuscher and H. Ji, Chem. Sci., 2016, 7, 2111–2118 RSC; (k) A. C. Edwards, A. Geist, U. Müllich, C. A. Sharrad, R. G. Pritchard, R. C. Whitehead and L. M. Harwood, Chem. Commun., 2017, 53, 8160–8163 RSC.
- (a) C. Raviola, S. Protti, D. Ravelli and M. Fagnoni, Green Chem., 2019, 21, 748–764 RSC; (b) D. Ravelli, S. Montanaro, M. Zema, M. Fagnoni and A. Albini, Adv. Synth. Catal., 2011, 353, 3295–3300 CrossRef CAS; (c) G. Bencivenni, T. Lanza, R. Leardini, M. Minozzi, D. Nanni, P. Spagnolo and P. Zanardi, J. Org. Chem., 2008, 73, 4721–4724 CrossRef CAS PubMed; (d) A. G. M. Barrett, H. Kwonband and E. M. Wallace, J. Chem. Soc., Chem. Commun., 1993, 1760–1761 RSC; (e) V. G. Correia, J. C. Abreu, C. A. E. Barata and L. H. Andrade, Org. Lett., 2017, 19, 1060–1063 CrossRef CAS PubMed; (f) M. B. Zhou, R.-J. Song, X.-H. Ouyang, Y. Liu, W.-T. Wei, G.-B. Deng and J.-H. Li, Chem. Sci., 2013, 4, 2690–2694 RSC; (g) M. Li, C. Wang, P. Fang and H. Ge, Chem. Commun., 2011, 47, 6587–6589 RSC; (h) W.-Z. Bi, W.-J. Zhang, Z.-J. Li, Y.-H. He, S.-X. Feng, Y. Geng, X.-L. Chene and L.-B. Qu, Org. Biomol. Chem., 2021, 19, 8701–8705 RSC; (i) M. Gautier, I. Rico and A. Lattes, J. Fluorine Chem., 1989, 44, 419–432 CrossRef CAS.
- M. Sakamoto, M. Takahashi, T. Fujita, T. Nishio, I. Iida and S. Watanabe, J. Org. Chem., 1995, 60, 4682–4683 CrossRef CAS.
- (a) G. Lopez-Valdez, S. Olguin-Uribe and L. D. Miranda, Tetrahedron Lett., 2007, 48, 8285–8289 CrossRef CAS; (b) A. Millan-Ortiz, G. Lopez-Valdez, F. Cortez-Guzman and L. D. Miranda, Chem. Commun., 2015, 51, 8345–8348 RSC; (c) G. Lopez-Valdez, S. Olguín-Uribe, A. Millan-Ortíz, R. Gamez-Montano and L. D. Miranda, Tetrahedron, 2011, 67, 2693–2701 CrossRef CAS; (d) M. Betou, L. Male, J. W. Steed and R. S. Grainger, Chem. – Eur. J., 2014, 20, 6505–6517 CrossRef CAS PubMed.
- (a) A. F. Bella, L. V. Jackson and J. C. Walton, Org. Biomol. Chem., 2004, 2, 421–428 RSC; (b) L. V. Jackson and J. C. Walton, Chem. Commun., 2000, 2327–2328 RSC; (c) S. B. Herzon and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 5342–5344 CrossRef CAS PubMed.
- S. Fujiwara, Y. Shimizu, T. Shin-ike and N. Kambe, Org. Lett., 2001, 3, 2085–2088 CrossRef CAS PubMed.
- P. H. Mazzocchi and M. Bowen, J. Org. Chem., 1976, 41, 1279–1282 CrossRef CAS.
- S. Maiti, S. Roy, P. Ghosh and D. Maiti, Chemrxiv, 2022 DOI:10.26434/chemrxiv-2021-1klwq.
- (a) E. de, P. Beato, D. Mazzarella, M. Balletti and P. Melchiorre, Chem. Sci., 2020, 11, 6312–6324 RSC; (b) N. Alandini, L. Buzzetti, G. Favi, T. Schulte, L. Candish, K. D. Collins and P. Melchiorre, Angew. Chem., Int. Ed., 2020, 59, 5248–5253 CrossRef CAS PubMed; (c) B. T. Matsuo, P. H. R. Oliveira, J. T. M. Correia and M. W. Paixão, Org. Lett., 2021, 23, 6775–6779 CrossRef CAS PubMed; (d) L. Cardinale, M. O. Konev and A. Jacobi von Wangelin, Chem. – Eur. J., 2020, 26, 8239–8243 CrossRef CAS PubMed; (e) L. Cardinale, M. O. W. S. Schmotz, M. O. Konev and A. Jacobi von Wangelin, Org. Lett., 2022, 24(2), 506–510 CrossRef CAS PubMed.
- (a) M. Yuan, L. Chen, J. Wang, S. Chen, K. Wang, Y. Xue, G. Yao, Z. Luo and Y. Zhang, Org. Lett., 2015, 17, 346–349 CrossRef CAS PubMed; (b) W. F. Petersen, R. J. K. Taylor and J. R. Donald, Org. Biomol. Chem., 2017, 15, 5831–5845 RSC.
- X. Chu, Y. Wu, H. Lu, B. Yang and C. Ma, Eur. J. Org. Chem., 2020, 1141–1144 CrossRef CAS.
- (a) M. Chmielewski, J. N. BeMiller and D. P. J. Cerretti, J. Org. Chem., 1981, 46, 3903–3908 CrossRef CAS; (b) M. B. Zhou, R.-J. Song, X.-H. Ouyang, Y. Liu, W.-T. Wei, G.-B. Deng and J.-H. Li, Chem. Sci., 2013, 4, 2690–2694 RSC.
- G. G. Pawar, F. Robert, E. Grau, H. Cramail and Y. Landais, Chem. Commun., 2018, 54, 9337–9340 RSC.
- (a) F. Minisci, E. Vismara and F. Fontana, Heterocycles, 1989, 28, 489–519 CrossRef CAS; (b) F. Minisci, F. Fontana and E. Vismara, J. Heterocycl. Chem., 1990, 27, 79–96 CrossRef CAS; (c) F. Coppa, F. Fontana, E. Lazzarini and F. Minisci, Heterocycles, 1993, 36, 2687–2696 CrossRef CAS.
- (a) J. Kos, C. F. Ku, I. Kapustikova, M. Oravec, H. J. Zhang and J. Jampilek, ChemistrySelect, 2019, 4, 4582–4587 CrossRef CAS; (b) J.-W. Yuan, Q. Chen, C. Li, J.-L. Zhu, L.-R. Yang, S.-R. Zhang, P. Mao, Y.-M. Xiao and L.-B. Qu, Org. Biomol. Chem., 2020, 18, 2747–2757 RSC.
- J.-W. Yuan, J.-L. Zhu, H.-L. Zhu, F. Peng, L.-Y. Yang, P. Mao, S.-R. Zhang, Y.-C. Lib and L.-B. Qu, Org. Chem. Front., 2020, 7, 273–285 RSC.
- (a) M. T. Westwood, C. J. C. Lamb, D. R. Sutherland and A.-L. Lee, Org. Lett., 2019, 21, 7119–7123 CrossRef CAS PubMed; (b) T. Mooney, B. D. T. Donkin, N. Demirel, P. R. Moore and A.-L. Lee, J. Org. Chem., 2021, 86, 17282–17293 CrossRef PubMed.
- V. S. Bhat and A. Lee, Eur. J. Org. Chem., 2021, 3382–3385 CrossRef CAS.
- A. H. Jatoi, G. G. Pawar, F. Robert and Y. Landais, Chem. Commun., 2019, 55, 466–469 RSC.
- S.-S. Zhu, Y. Liu, X.-L. Chen, L.-B. Qu and B. Yu, ACS Catal., 2022, 12, 126–134 CrossRef CAS.
- M. Jouffroy and J. Kong, Chem. – Eur. J., 2019, 25, 2217–2221 CrossRef CAS PubMed.
- X.-L. Lai, X.-M. Shu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS PubMed.
- K. Jing, P.-C. Cui and G.-W. Wang, Chem. Commun., 2019, 55, 12551–12554 RSC.
- W.-M. Cheng, R. Shang, H.-Z. Yu and Y. Fu, Chem. – Eur. J., 2015, 21, 13191–13195 CrossRef CAS PubMed.
- Q. Jiang, J. Jia, B. Xu, A. Zhao and C.-C. Guo, J. Org. Chem., 2015, 80, 3586–3596 CrossRef CAS PubMed.
- H. Wang, L.-N. Guo, S. Wang and X.-H. Duan, Org. Lett., 2015, 17, 3054–3057 CrossRef CAS PubMed.
- H. Huang, G. Zhang and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 7872–7876 CrossRef CAS PubMed.
- M. Yuan, L. Chen, J. Wang, S. Chen, K. Wang, Y. Xue, G. Yao, Z. Luo and Y. Zhang, Org. Lett., 2015, 17, 346–349 CrossRef CAS PubMed.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018–5022 CrossRef CAS PubMed.
- H. Fan, P. Pan, Y. Zhang and W. Wang, Org. Lett., 2018, 20, 7929–7932 CrossRef CAS PubMed.
- V. V. Zhdankin, A. E. Koposov and J. T. Smart, J. Am. Chem. Soc., 2001, 123, 4095–4096 CrossRef CAS PubMed.
- Q.-Q. Han, Y.-Y. Sun, S.-H. Yang, J.-C. Song and Z.-L. Wang, Chin. Chem. Lett., 2021, 32, 3632–3635 CrossRef CAS.
- Z. Zhang, C. Jia, X. Kong, M. Hussain, Z. Liu, W. Liang, L. Jiang, H. Jiang and J. Ma, ACS Sustainable Chem. Eng., 2020, 8, 16463–16468 CrossRef CAS.
- Q. Liu, L. Wang, J. Liu, S. Ruan and P. Li, Org. Biomol. Chem., 2021, 19, 3489–3496 RSC.
- G. Chen, C. Li, J. Peng, Z. Yuan, P. Liu and X. Liu, Org. Biomol. Chem., 2019, 17, 8527–8532 RSC.
- C. Jin, J.-Y. He, Q.-F. Bai and G. Feng, Synlett, 2020, 1517–1522 CAS.
- Q.-F. Bai, C. Jin, J.-Y. He and G. Feng, Org. Lett., 2018, 20, 2172–2175 CrossRef CAS PubMed.
- C. M. Mazodze and W. F. Petersen, Org. Biomol. Chem., 2022, 20, 3469–3474 RSC.
- L. Wang, H. Wang, Y. Wang, M. Shen and S. Li, Tetrahedron Lett., 2020, 61, 151962 CrossRef CAS.
- I. M. Ogbu, J. Lusseau, G. Kurtay, F. Robert and Y. Landais, Chem. Commun., 2020, 56, 12226–12229 RSC.
- A. Petti, C. Fagnan, C. G. W. van Melis, N. Tanbouza, A. D. Garcia, A. Mastrodonato, M. C. Leech, I. C. A. Goodall, A. P. Dobbs, T. Ollevier and K. Lam, Org. Process Res. Dev., 2021, 25, 2614–2621 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |