
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoelectronically-induced allosteric binding: shape complementarity promotes positive cooperativity in fullerene/buckybowl complexes†
Eric S.
Larsen
 ab,
Guillermo
Ahumada
a,
Prakash R.
Sultane
a and
Christopher W.
Bielawski
*ab
ab,
Guillermo
Ahumada
a,
Prakash R.
Sultane
a and
Christopher W.
Bielawski
*ab
aCenter for Multidimensional Carbon Materials (CMCM), Institute for Basic Science (IBS), Ulsan, 44919, Republic of Korea. E-mail: bielawski@unist.ac.kr
bDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan, 44919, Republic of Korea
First published on 16th May 2022
Abstract
A novel 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest complex forms between 8-tert-butyl-6b2-azapenta-benzo[bc,ef,hi,kl,no]corannulene (1) and C60 with positive cooperativity (α = 2.56) and high affinity (K1 × K2 = 2.8 × 106 M−2) at 25 °C. The C60 undergoes increasing shape complementarity toward 1 throughout the binding process.
:1 host–guest complex forms between 8-tert-butyl-6b2-azapenta-benzo[bc,ef,hi,kl,no]corannulene (1) and C60 with positive cooperativity (α = 2.56) and high affinity (K1 × K2 = 2.8 × 106 M−2) at 25 °C. The C60 undergoes increasing shape complementarity toward 1 throughout the binding process.
The discovery of the fullerenes1 prompted efforts toward finding applications for the novel carbon allotrope, ranging from photovoltaics,2,3 and organic electronics4 to medicine5,6 and self-assembly.7 A landmark demonstration8 of the latter was reported by Wennerström who showed that a host–guest complex forms between C60 and γ-cyclodextrin. Since then, the field9–11 has progressed toward achieving selective fullerene complexation to not only expedite isolation and purification12–15 but to also facilitate the construction of organized nanostructures.16–20 The formation of stable assemblies is apropos to achieving these aims and a broad variety of molecular components have been explored for their abilities to form complexes with fullerenes, including calixarenes,21 tetrathiafulvalenes,22,23 subphthalocyanines,24 and porphyrins.25,26
Due to the complementarity of their respective concave and convex surfaces, bowl-shaped polycyclic aromatic hydrocarbons (PAHs) – often colloquially referred to as “buckybowls” – are prime candidates for forming assemblies with C60.25,27–29 For example, corannulene,30 a prototypical buckybowl, has been shown to form a 1:1 complex with C60 that resembles a ball-and-socket joint (Fig. 1), albeit only in the solid state.31,32 To enhance buckybowl-fullerene interactions, PAHs have been enlarged to increase surface area and/or modified with electron-rich components.27 For example, association constant (Ka) values of up to 1400 M−1 were measured in solution (toluene-d8) when corannulene was decorated with pendant arylthiol groups and then treated with C60.33,34 An even larger association constant was measured (6.2 × 104 M−1, toluene) when corannulene was integrated with an electron-rich heteroatom (i.e., azacorannulene) and then introduced to electron-deficient C60.35–38
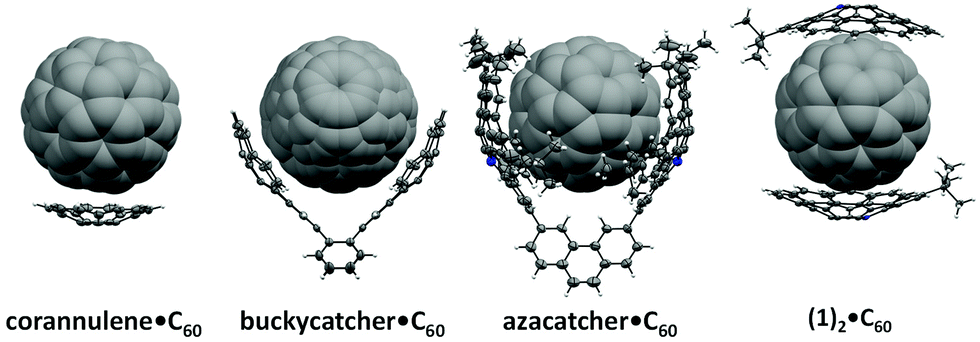 | ||
| Fig. 1 Structures of selected buckybowl·C60 complexes. | ||
Binding enhancement has also been demonstrated by tethering multiple buckybowls in such a way that they become preorganized to chelate C60.27 For example, the “buckycatcher” consists of two corannulene units spanned across a tetrabenzocyclooctatetraene linker and acts as a type of molecular tweezer for C60 (Ka = 2.7 × 103 M−1, toluene-d8).39,40 Azacorannulene-based tweezers have also been synthesized and shown to display large association constants with C60 (up to 3.0 × 108 M−1 in toluene), depending on the linker used.38 Despite the growing diversity, the majority of buckybowl-fullerene complexes can be classified as 1:1 host–guest systems. Multi-component assemblies comprised of multiple fullerene hosts and/or multiple buckybowls are relatively rare in solid21,41–46 or solution states,28,47,48 and typically utilize modified buckybowls or porphyrins. “Super-stoichiometric” complexes are attractive because they can be expected to exhibit relatively high stabilities and thus form with greater selectivity when compared to their stoichiometric analogues.49,50
An alternative approach to forming super-stoichiometric assemblies between buckybowls and fullerenes was envisioned. Although buckybowls generally feature smaller curvatures when compared to that of C60, the binding affinity between two juxtaposed surfaces can be expected to increase as their curvatures become better matched. Moreover, complexation of an electron-rich buckybowl to C60 should result in charge transfer that effectively widens the curvature of the uncomplexed area of the fullerene. Such a process may increase the topological complementarity between the complexed C60 intermediate and a free buckybowl,51 and thus may favor the formation of a super-stoichiometric complex. To test this hypothesis, an azacorannulene (1) which features a surface that is curved, electron-rich and mono-substituted to facilitate binding with C60 while maintaining a reasonably high degree of solubility was selected.37 As will be described below, it was discovered that the azacorannulene not only forms a 2:1 complex with C60 in solution and in the solid-state but also binds to C60 with positive cooperativity as predicted by the hypothesis. Variable temperature titrations, calculations and structural models are also presented to gain a deeper understanding of the complex that formed and to clarify the unique binding phenomena. In a broader context, the discovery constitutes a new method for using allosteric changes to form super-stoichiometric complexes with fullerenes.
The intermolecular interactions formed between 1, which was synthesized by modifying a literature37 procedure (see ESI,† Scheme S1), and C60 were first probed through a series of titrations and monitored via1H NMR spectroscopy. Treating a solution of the buckybowl ([1]0 = 240 μM in toluene-d8) with increasing quantities of C60 ([C60]0 = 2.5 mM) generally caused the 1H NMR signals that were assigned to 1 to shift upfield although the signals assigned to the tert-butyl group and to the hydrogen atom located at the 2-position shifted downfield (Fig. 2a). The former chemical shifts can be attributed to shielding effects (e.g., CH–π interactions), while the latter may be induced via interactions with the LUMO on C60 (vide infra). The systematic shift in signals indicated that a well-defined complex formed in situ as the two components were combined.52
 | ||
| Fig. 2 (a) 1H NMR spectra that were recorded as 1 (240 μM) was titrated with increasing quantities of C60 (2.5 mM) (indicated) in toluene-d8 at 25 °C. (b) Job Plot used to determine the stoichiometry of the complex formed as obtained by measuring the response, defined as |Δδ| × 106, between 1 and C60 in toluene-d8 at 25 °C. (c) Averaged binding isotherm data (black circles) with a 2:1 cubic fit (blue line) of the hydrogen assigned to the 2-position of 1; conditions: [1]0 = 240 μM, [C60]0 = 2.5 mM, toluene-d8, 25 °C. (d) ORTEP diagram of (1)2·C60. Thermal displacement ellipsoids are drawn at the 50% probability level. | ||
A Job plot was constructed to determine the stoichiometry of the complex formed. A maximum was observed when the mole fraction of C60 was approximately one-third, consistent with the formation of a 2:1 complex (Fig. 2b).53,54 To verify the stoichiometry and to measure the constituent association constants, binding isotherm data were plotted and analyzed with BindFit,55 a non-linear regression analysis program. The binding isotherm featured an inflection point when the stoichiometry of the two components was 2:1 and then began to saturate (Fig. 2c). Likewise, good agreement between the binding isotherm data and a cubic fit that follows a 2:1 model (i.e., (1)2·C60) was obtained56,57 and the association constants for the two binding events were determined to be K1 = 2126 ± 87 M−1 and K2 = 1359 ± 103 M−1. The first binding event appeared to facilitate the second event as a cooperativity factor (α), defined as 4K2/K1, of 2.56 was calculated for the system.55 The overall affinity of 1 toward C60 can be calculated (K1 × K2 = 2.8 × 106 M−2)55,58,59 and compared to the values reported for the aforementioned molecular tweezers.38 For reference, the self-association constants of 1 (2.4 ± 0.4 M−1)37–60 and (1)2·C60 (10.7 ± 2.9 M−1) were measured in toluene-d8 at 25 °C and determined to be relatively small. Collectively, the data indicated that a tight 2:1 complex formed between 1 and C60 and that the formation process occurred selectively and with positive cooperativity.
A van’t Hoff analysis was performed to gain a deeper insight into the process that governs complex formation. The analysis indicated that the first binding event (i.e., the formation of 1·C60) is enthalpy-driven and entropically disfavored (ΔH = −5.3 kcal mol−1; ΔS = −3.0 cal mol−1 K−1) which may be due in part to a loss in translational freedom. However, the intermediate complex (i.e., 1·C60) appears to be preorganized toward binding additional 1 as the second binding event (i.e., the formation of (1)2·C60) was measured to be entropically favored (ΔS = +16.0 cal mol−1 K−1) in accord with the hypothesis. The second step appears to incur an enthalpic penalty (ΔH = +0.65 kcal mol−1) which may stem from a repulsive electrostatic interaction as the C60 component in the intermediate complex should be relatively electron-rich due to charge transfer. Support for this conclusion was obtained via cyclic voltammetry which revealed that (1)2·C60 exhibited a lower reduction potential than the C60 host (Ered = −1.03 V vs. −1.00 V, respectively, in 5:1 v/v o-dichlorobenzene/acetonitrile) as well as a higher oxidation potential than its constituent guest (1) (Eox = +0.37 V vs. +0.35 V, respectively). Additional support was obtained from the structural analyses and the calculations described below.
Further evidence that supported the formation of a 2:1 complex between 1 and C60 was obtained by a single-crystal X-ray diffraction analysis. The solid-state structure features a concave-convex relationship analogous to those observed in other types of buckybowl·fullerene complexes reported in the literature (Fig. 2d).31,33,35 The bowl-to-ball (BtB) distance measured for (1)2·C60 (6.88 Å) was intermediate of the values reported for the solid-state structures of (2,5,11,14-tetra-tert-butyl-6b2-azapentabenzo[bc,ef,hi,kl,no]-corannulene)·C60 (6.82 Å)35 and corannulene·C60 (6.94 Å).31 The assembly angle (θ), which is defined as the angle between the center of C60 and the pyrrole centroids on 1, was measured to be 180.0°, indicating that the two guests were antipodal. Inspection of the X-ray data revealed that every ring in 1 was within 4.0 Å of C60, consistent with a π–π interaction,61 and that the number of π–π contacts formed appeared to depend on the depth of the buckybowl. For example, while only one contact between the central ring of 1 and C60 was identified, an average of 1.6 and 1.4 contacts to C60 were measured for the inner and peripheral rings, respectively. A series of CH–π interactions in the crystal lattice were also identified and may serve to stabilize the crystal lattice or to guide packing (see ESI,† Fig. S50–S52).
A series of structural analyses were performed to assess shape complementarity and a summary is shown in Fig. 3.62 Although a π-orbital axis vector63,64 (POAV) angle of 8.28° has been reported for 1,37 the average POAV angle of the pyrrolyl carbons in (1)2·C60 was wider (8.37°) and thus better matched with the angle reported for C60 (11.64°).65 The remaining carbon atoms in the inner rings of the complex also displayed a larger average hybridization angle (3.02°) than that of virgin 1 (2.88°). The POAV differential resulted in a deeper bowl (c.f., the distance from the pyrrole centroid to the outermost carbon atoms in the buckybowl) of (1)2·C60 (1.63 Å) when compared to that measured for 1 (1.58 Å).37 Likewise, the eccentricity (e) of 1 was also measured to decrease upon complexation. In other words, the azacorannulene guests appeared to have flexed to better match the topology of the C60 host. The binding phenomena effectively compressed C60 into a prolate spheroid as the semi-minor (binding) axis (e = 0.000) of the host was shorter than its semi-major axis (e = 0.024). The average POAV angle calculated for the binding regions was also relatively acute (11.55°) when compared to that of uncomplexed C60.65
 | ||
Fig. 3 Illustrations of the corresponding calculated changes in eccentricity  , curvature ( , curvature (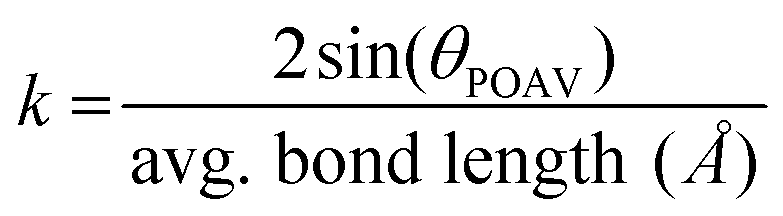 ) and POAV values at different regions of C60 (shaded blue) as obtained from the (a–c) DFT or (d) X-ray crystal data. The eccentricity, curvature and POAV values for 1 are 0.948, 0.204 Å−1 and 8.11°, respectively. The tert-butyl groups on 1 were omitted for clarity. ) and POAV values at different regions of C60 (shaded blue) as obtained from the (a–c) DFT or (d) X-ray crystal data. The eccentricity, curvature and POAV values for 1 are 0.948, 0.204 Å−1 and 8.11°, respectively. The tert-butyl groups on 1 were omitted for clarity. | ||
To obtain a deeper understanding of the supramolecular assembly process and the structures of the complexes that formed, a series of DFT calculations (M06-2X-D3/6-31G(d,p)) were conducted on C60, 1, 1·C60 and (1)2·C60. Good agreement between the X-ray data collected for (1)2·C60 and the calculated data was realized. For example, the calculated assembly angle of the azacorannulene units (179.9°) and the BtB distance (6.73 Å) were consistent with the X-ray data. Key intermolecular charge-transfer interactions were also identified upon inspection of the structures calculated for (1)2·C60 and 1·C60. The HOMO was found to reside on the azacorannulene units whereas the LUMO was located on C60. The calculations showed that the first binding event effectively caused the opposite (open) region of the host to experience compression as the semi-minor axis of C60 in 1·C60 (7.073 Å) was contracted when compared to that of virgin C60 (7.079 Å). A curvature value65 (k) of 0.282 Å−1 was calculated for the compressed region in 1·C60, which is more complementary to that measured for 1 (0.204 Å−1). For reference, k values of 0.284 Å−1 and 0.281 Å−1 were calculated for uncomplexed C60 and the C60 contained in (1)2·C60, respectively. Similarly, the POAV angle calculated for the open region in 1·C60 was reduced and thus better matched the surface topology of 1.
The subtleties of such stereoelectronically-induced, allosteric changes were borne out in the binding phenomena. As noted above, the K1 value was measured to be larger than the K2 value and the overall binding process was measured to proceed with positive cooperativity. Consistent with the experimental data, enthalpy of formation values of −31.8 and −30.8 kcal mol−1 were calculated for 1·C60 and (1)2·C60, respectively. The LUMO of 1·C60 (−2.49 eV) was slightly higher in energy than that of C60 (−2.70 eV), which may explain why the second binding event is less favored than the first. The exposed surface of 1·C60 is relatively electron-rich due to charge transfer and thus may disfavor binding to a second unit of (electron-rich) 1. However, the electronic offset caused by the first binding event appears to be compensated by steric changes that effectively increase the topological complementarity between the binding partners.
In summary, it was demonstrated that two units of azacorannulene 1 bind to C60 in solution as well as in the solid-state. The binding phenomena proceed with high affinity and positive cooperativity as determined by titration experiments and DFT calculations. The binding of 1 to C60 results in charge-transfer and causes the fullerene to adopt a compressed spheroid geometry. The stereoelectronic effect afforded an open site in the resulting 1·C60 intermediate that displayed increased shape complementarity to 1, as indicated by a series of POAV and curvature analyses, and ultimately facilitated the second binding event. The allosteric changes were corroborated with an X-ray analysis as well as by a series of thermodynamic measurements and calculations. In a broader perspective, the design principles described should faciliate access to super-stoichiometric carbon assemblies with potential applications in carbon chemistry, materials science, and nanotechnology.
The IBS (R01-019-D1) is acknowledged for support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- C. Z. Li, H. L. Yip and A. K.-Y. Jen, J. Mater. Chem., 2012, 22, 4161–4177 RSC.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- D. M. Guldi, B. M. Illescas, C. M. Atienza, M. Wielopolskia and N. Martin, Chem. Soc. Rev., 2009, 38, 1587–1597 RSC.
- E. Castro, A. H. Garcia, G. Zavala and L. Echegoyen, J. Mater. Chem. B, 2017, 5, 6523–6535 RSC.
- P. Anilkumar, F. Lu, L. Cao, P. G. Luo, J. H. Liu, S. Sahu, K. N. Tackett, Y. Wang and Y. P. Sun, Curr. Med. Chem., 2011, 18, 2045–2059 CrossRef CAS PubMed.
- F. Diederich and M. Gomez-Lopez, Chem. Soc. Rev., 1999, 28, 263–277 RSC.
- T. Andersson, K. Nilsson, M. Sundahl, G. Westman and O. Wennerstrom, J. Chem. Soc., Chem. Commun., 1992, 604–606 RSC.
- N. Komatsu, J. Inclusion Phenom. Macrocyclic Chem., 2008, 61, 195–216 CrossRef CAS.
- S. S. Babu, H. Mohwald and T. Nakanishi, Chem. Soc. Rev., 2010, 39, 4021–4035 RSC.
- T. Nakanishi, Chem. Commun., 2010, 46, 3425–3436 RSC.
- Y. Shi, K. Cai, H. Xiao, Z. C. Liu, J. W. Zhou, D. K. Shen, Y. Y. Qiu, Q. H. Guo, C. Stern, M. R. Wasielewski, F. Diederich, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 13835–13842 CrossRef CAS PubMed.
- C. Garcia-Simon, M. Garcia-Borras, L. Gomez, T. Parella, S. Osuna, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Nat. Commun., 2014, 5 Search PubMed.
- K. Nagata, E. Dejima, Y. Kikuchi and M. Hashiguchi, Chem. Lett., 2005, 34, 178–179 CrossRef CAS.
- Y. Shoji, K. Tashiro and T. Aida, J. Am. Chem. Soc., 2010, 132, 5928–5929 CrossRef CAS PubMed.
- T. Haino, E. Hirai, Y. Fujiwara and K. Kashihara, Angew. Chem., Int. Ed., 2010, 49, 7899–7903 CrossRef CAS PubMed.
- X. Zhang and M. Takeuchi, Angew. Chem., Int. Ed., 2009, 48, 9646–9651 CrossRef CAS PubMed.
- G. Fernandez, E. M. Perez, L. Sanchez and N. Martin, J. Am. Chem. Soc., 2008, 130, 2410–2411 CrossRef CAS PubMed.
- S. Barrau, T. Heiser, F. Richard, C. Brochon, C. Ngov, K. van de Wetering, G. Hadziioannou, D. V. Anokhin and D. A. Ivanov, Macromolecules, 2008, 41, 2701–2710 CrossRef CAS.
- M. Schmittel, B. He and P. Mal, Org. Lett., 2008, 10, 2513–2516 CrossRef CAS PubMed.
- T. Haino, M. Yanase and Y. Fukazawa, Angew. Chem., Int. Ed. Engl., 1997, 36, 259–260 CrossRef CAS.
- S. Goeb and M. Salle, Acc. Chem. Res., 2021, 54, 1043–1055 CrossRef CAS PubMed.
- D. Canevet, M. Gallego, H. Isla, A. de Juan, E. M. Perez and N. Martin, J. Am. Chem. Soc., 2011, 133, 3184–3190 CrossRef CAS PubMed.
- R. Ziessel, G. Ulrich, K. J. Elliott and A. Harriman, Chem. – Eur. J., 2009, 15, 4980–4984 CrossRef CAS PubMed.
- Y. T. Wu and J. S. Siegel, Top. Curr. Chem., 2014, 349, 63–120 CrossRef CAS PubMed.
- J. Y. Zheng, K. Tashiro, Y. Hirabayashi, K. Kinbara, K. Saigo, T. Aida, S. Sakamoto and K. Yamaguchi, Angew. Chem., Int. Ed., 2001, 40, 1858–1861 Search PubMed.
- T. Kawase and H. Kurata, Chem. Rev., 2006, 106, 5250–5273 CrossRef CAS PubMed.
- M. Yanney, F. R. Fronczek and A. Sygula, Angew. Chem., Int. Ed., 2015, 54, 11153–11156 CrossRef CAS PubMed.
- J. W. Steed, P. C. Junk, J. L. Atwood, M. J. Barnes, C. L. Raston and R. S. Burkhalter, J. Am. Chem. Soc., 1994, 116, 10346–10347 CrossRef CAS.
- W. E. Barth and R. G. Lawton, J. Am. Chem. Soc., 1971, 93, 1730–1745 CrossRef.
- L. N. Dawe, T. A. AlHujran, H. A. Tran, J. I. Mercer, E. A. Jackson, L. T. Scotc and P. E. Georghiou, Chem. Commun., 2012, 48, 5563–5565 RSC.
- W. Xiao, D. Passerone, P. Ruffieux, K. Ait-Mansour, O. Groning, E. Tosatti, J. S. Siegel and R. Fasel, J. Am. Chem. Soc., 2008, 130, 4767–4771 CrossRef CAS PubMed.
- P. E. Georghiou, A. H. Tran, S. Mizyed, M. Bancu and L. T. Scott, J. Org. Chem., 2005, 70, 6158–6163 CrossRef CAS PubMed.
- S. Mizyed, P. E. Georghiou, M. Bancu, B. Cuadra, A. K. Rai, P. C. Cheng and L. T. Scott, J. Am. Chem. Soc., 2001, 123, 12770–12774 CrossRef CAS PubMed.
- H. Yokoi, Y. Hiraoka, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- M. Saito, H. Shinokubo and H. Sakurai, Mater. Chem. Front., 2018, 2, 635–661 RSC.
- S. Ito, Y. Tokimaru and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 7256–7260 CrossRef CAS PubMed.
- M. Takeda, S. Hiroto, H. Yokoi, S. Lee, D. Kim and H. Shinokubo, J. Am. Chem. Soc., 2018, 140, 6336–6342 CrossRef CAS PubMed.
- A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842–3843 CrossRef CAS PubMed.
- V. H. Le, M. Yanney, M. McGuire, A. Sygula and E. A. Lewis, J. Phys. Chem. B, 2014, 118, 11956–11964 CrossRef CAS PubMed.
- M. M. Olmstead, D. A. Costa, K. Maitra, B. C. Noll, S. L. Phillips, P. M. Van Calcar and A. L. Balch, J. Am. Chem. Soc., 1999, 121, 7090–7097 CrossRef CAS.
- X. S. Ke, T. Kim, J. T. Brewster, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2017, 139, 4627–4630 CrossRef CAS PubMed.
- Y. H. Yang, K. M. Cheng, Y. Lu, D. D. Ma, D. H. Shi, Y. X. Sun, M. Y. Yang, J. Li and J. F. Wei, Org. Lett., 2018, 20, 2138–2142 CrossRef CAS PubMed.
- Y. Y. Xu, H. R. Tian, S. H. Li, Z. C. Chen, Y. R. Yao, S. S. Wang, X. Zhang, Z. Z. Zhu, S. L. Deng, Q. Y. Zhang, S. F. Yang, S. Y. Xie, R. B. Huang and L. S. Zheng, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- A. S. Filatov, M. V. Ferguson, S. N. Spisak, B. Li, C. F. Campana and M. A. Petrukhina, Cryst. Growth Des., 2014, 14, 756–762 CrossRef CAS.
- H. Yokoi, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Chem. Sci., 2018, 9, 819–824 RSC.
- Y. Saegusa, T. Ishizuka, T. Kojima, S. Mori, M. Kawano and T. Kojima, Chem. – Eur. J., 2015, 21, 5302–5306 CrossRef CAS PubMed.
- P. L.-A. Kuragama, F. R. Fronczek and A. Sygula, Org. Lett., 2015, 17, 5292–5295 CrossRef PubMed.
- F. Hof, S. L. Craig, C. Nuckolls and J. Rebek, Angew. Chem., Int. Ed., 2002, 41, 1488–1508 CrossRef CAS PubMed.
- J. Rebek, Acc. Chem. Res., 1984, 17, 258–264 CrossRef CAS.
- A. S. Filatov, E. A. Jackson, L. T. Scott and M. A. Petrukhina, Angew. Chem., Int. Ed., 2009, 48, 8473–8476 CrossRef CAS PubMed.
- Isosbestic points were observed at 425 and 625 nm upon titrating C60 with 1 and provided further support that a well-defined complex was formed (see ESI,† Fig. S35).
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- F. Ulatowski, K. Dabrowa, T. Balakier and J. Jurczak, J. Org. Chem., 2016, 81, 1746–1756 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 5922–5923 RSC.
- The differences between the model and the recorded data were plotted and found to be small (see ESI,† Fig. S31).
- Consistent with complexation, the diffusion coefficient of (1)2·C60 (6.54 × 10−10 m2 s−1) was smaller than that of 1 (7.43 × 10−10 m2 s−1) (see ESI,† Fig. S36, S37 and Table S9).
- K. A. Connors, Binding constants: the measurement of molecular complex stability, Wiley, New York, 1987 Search PubMed.
- L. K.-S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC.
- A similar self-association constant (5.0 ± 0.4 M−1) was reported37 for 1 in deuterated benzene at 25 °C.
- C. A. Hunter and J. K.-M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- More details are in the ESI;† see Fig. S53–S58 and Tables S17–S23.
- R. C. Haddon, J. Phys. Chem. A, 2001, 105, 4164–4165 CrossRef CAS.
- R. C. Haddon, J. Am. Chem. Soc., 1986, 108, 2837–2842 CrossRef CAS.
- R. C. Haddon, J. Am. Chem. Soc., 1997, 119, 1797–1798 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures; additional titration data and binding isotherms; UV-vis and NMR spectra; cyclic voltammograms; X-ray crystallographic data for (1)2·C60; additional geometric measurements; DFT data. CCDC 2115565. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc01908f |
| This journal is © The Royal Society of Chemistry 2022 |