
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly flexible phosphabenzenes: a missing coordination mode of 2,4,6-triaryl-λ3-phosphinines†‡
Erlin
Yue§
 ab,
Andrey
Petrov§
a,
Daniel S.
Frost
a,
Lea
Dettling
a,
Lawrence
Conrad
a,
Friedrich
Wossidlo
a,
Nathan T.
Coles
a,
Manuela
Weber
a and
Christian
Müller
*a
ab,
Andrey
Petrov§
a,
Daniel S.
Frost
a,
Lea
Dettling
a,
Lawrence
Conrad
a,
Friedrich
Wossidlo
a,
Nathan T.
Coles
a,
Manuela
Weber
a and
Christian
Müller
*a
aFreie Universität Berlin, Institute of Chemistry and Biochemistry, Fabeckstr. 34/36, 14195 Berlin, Germany. E-mail: c.mueller@fu-berlin.de
bShaanxi Key Laboratory of Chemical Reaction Engineering, Key Laboratory of New Energy & New Functional Materials, College of Chemistry and Chemical Engineering, Yan'an University, Yan'an, Shaanxi 716000, People's Republic of China. E-mail: yueerlin@yau.edu.cn
First published on 13th April 2022
Abstract
The reaction of 2,4,6-triaryl-λ3-phosphinine-Cr(CO)3-π-complexes with [Rh(COD)2]BF4 leads to unusual diamagnetic Rh0-dimers, which contain two phosphinine-π-complexes acting as a bridging 2e−-ligand towards the Rh2(CO)2 core. These compounds represent a missing coordination mode for the aromatic 6-membered phosphorus heterocycle.
In contrast to classical phosphines (PR3), phosphinines (phosphabenzenes) show rich and diverse coordination chemistry.1 As ambidentate ligands, these aromatic phosphorus heterocycles can adopt various coordination modes, in which the phosphorus atom and/or the π-system is involved in the interaction with the metal centre (Chart 1).
 | ||
| Chart 1 Coordination modes of λ3,σ2-phosphinines. | ||
The fascinating coordination chemistry of phosphinines has been explored extensively during the last two decades, with particular focus on the reactivity of transition metal phosphinine complexes and their application in homogeneous catalysis and materials science.2
η1-Phosphinine complexes (A, 2e−-donor) are common and are typically formed with transition metals in low oxidation states.1 This is due to the fact that phosphinines are very good π-acceptors when the coordination occurs via the phosphorus atom.3 Transition metal complexes with phosphinines as bridging ligands are also known (B, μ2-P, 2e−-donor).1 More recently, it has been shown that electron rich phosphinines (B, R = O−, NMe2) have a high tendency to act as 4e−, μ2-bridging ligands.4,5 It is also possible to coordinate via the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (C, η1-P, η2-PC, 4e−-donor).1 Phosphinine-η6-complexes (D, 6e−-donor) are also accessible,1 usually by addition of sterically demanding substituents, such as t-butyl- or Me3Si-groups in the α-position of the phosphorus heterocycle, which can prevent σ-coordination via the phosphorus lone-pair.6 With electron rich metal centers, coordination with the π-system can also occur in an η4-fashion (η2-CC, 4e−-donor) to fulfill the 18e− rule.1
C double bond (C, η1-P, η2-PC, 4e−-donor).1 Phosphinine-η6-complexes (D, 6e−-donor) are also accessible,1 usually by addition of sterically demanding substituents, such as t-butyl- or Me3Si-groups in the α-position of the phosphorus heterocycle, which can prevent σ-coordination via the phosphorus lone-pair.6 With electron rich metal centers, coordination with the π-system can also occur in an η4-fashion (η2-CC, 4e−-donor) to fulfill the 18e− rule.1
Examples, in which the η6-π-complex D serves as a 2e− metallo-ligand (E, η6-PC5, η1-P, 8e−-donor) are surprisingly rare.7 On the other hand, such bimetallic species may show interesting properties, which could be exploited in more applied research fields. The corresponding coordination compound F, in which the metallo-ligand D acts as a bridging 2e−-donor (η6-PC5, μ2-P) has so far not been reported in the literature. Elschenbroich and co-workers described the related species G, in which the Mn(CO)3-η5-phosphinine complex coordinates to a binuclear Mn2(CO)7 fragment.8 In this compound, the phosphorus atom thus serves as a 3e−-donor, while all Mn0-atoms fulfill the 18 electron rule.
Motivated by the lack of information on complexes of type E, we started to investigate the electronic properties and the coordination chemistry of the metallo-ligand D in more detail and report the first example of the missing coordination mode η6-PC5-μ2 (8e−) for phosphinines. As arene-η6-Cr(CO)3 complexes are very common organometallic 18 VE-species, we anticipated that the corresponding phosphinine complexes would be a good starting point for acquiring these species. The π-complex 2,4,6-triphenyl-λ3-phosphinine-Cr(CO)3 (2a) has been reported and structurally characterized by Deberitz and Nöth in 1970.9 The derivatives 2b/c are easily accessible in moderate yields from 2,4,6-triarylphosphinines 1b/c (Scheme 1). The π-complex 2a shows a single resonance in the 31P{1H} NMR spectrum at δ(ppm) = 6.1 (2b: δ(ppm) = 5.5; 2c: δ(ppm) = 5.1; Fig. S1, S5 and S8, ESI‡).
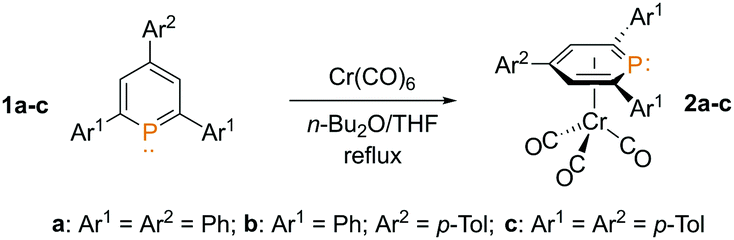 | ||
| Scheme 1 Synthesis of phosphinine-Cr(CO)3-π-complexes 2a–2c. | ||
We were first interested in the electronic properties of the phosphinine-π-complexes. Fig. S53 (ESI‡) illustrates the frontier Kohn–Sham-orbitals of the π-complex (C5H5P)Cr(CO)3 (2a′) in comparison to the parent phosphinine C5H5P. In general, phosphinines are good π-acceptors due to an energetically low-lying LUMO (Fig. S53, ESI‡, left side). On the other hand, they possess weak σ-donor properties, as the phosphorus-lone pair is energetically stabilized (HOMO−2). Interestingly, the corresponding Cr(CO)3-π-complex is both a poorer π-acceptor and an even weaker σ-donor than C5H5P (Fig. S53, ESI‡, right side). Thus, it is not surprising that relatively little is known about the coordination chemistry of phosphinine-π-complexes. We anticipated that Cr(CO)3-phosphinine-π-complexes might only be suitable as a metallo-ligand for the coordination to electron rich metal centers.
Because RhI complexes of phosphinines are usually straightforward to synthesize, we reacted 2a–c at room temperature with one equivalent of [Rh(COD)2]BF4 in CD2Cl2. Immediately, a single new species is formed, which shows a doublet resonance in the 31P{1H} NMR spectrum at δ(ppm) = 36.8 (1JP−Rh = 179 Hz, 3a) when using 2a as the ligand (3b: δ(ppm) = 36.0 (1JP−Rh = 179 Hz); 3c: δ(ppm) = 36.2 (1JP−Rh = 179 Hz); Fig. S11, S16 and S21, ESI‡). When the reaction is performed with a ligand![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :metal ratio of 2:1 (2a–c: Rh), one equivalent of 2a–c remains in solution. Attempts to crystalize the corresponding products 3a–c were unsuccessful. However, upon substituting the [BF4]− counterion of 2c for [BArF4]− (ArF = (CF3)2C6H3), we were able to isolate the RhI complex 3d, which was characterized crystallographically. The molecular structure of 3d in the crystal confirms the presence of two Cr(CO)3-phosphinine-π-complexes bound to the [Rh(COD)]+ fragment via the phosphorus lone-pair (Fig. 1). To confirm whether the observed structure of 3a–c was retained in solution, the 31P{1H} NMR spectrum of a reaction containing both 2a and 2b with [Rh(COD)2]BF4 in a ratio of 1:1:1 gave signals for a species with an ABX spin system, indicating the formation of [Rh(2a)(2b)(COD)]BF4 (Fig. S26, ESI‡). The spectrum also reveals two doublets and two singlets that correspond to 3a and 3b, as well as to free ligands 2a and 2b respectively. Based on this NMR experiment, we anticipate that two metallo-ligands are coordinated to one RhI-center to form 3a–c which are in a dynamic equilibrium in solution (Scheme 2).
:metal ratio of 2:1 (2a–c: Rh), one equivalent of 2a–c remains in solution. Attempts to crystalize the corresponding products 3a–c were unsuccessful. However, upon substituting the [BF4]− counterion of 2c for [BArF4]− (ArF = (CF3)2C6H3), we were able to isolate the RhI complex 3d, which was characterized crystallographically. The molecular structure of 3d in the crystal confirms the presence of two Cr(CO)3-phosphinine-π-complexes bound to the [Rh(COD)]+ fragment via the phosphorus lone-pair (Fig. 1). To confirm whether the observed structure of 3a–c was retained in solution, the 31P{1H} NMR spectrum of a reaction containing both 2a and 2b with [Rh(COD)2]BF4 in a ratio of 1:1:1 gave signals for a species with an ABX spin system, indicating the formation of [Rh(2a)(2b)(COD)]BF4 (Fig. S26, ESI‡). The spectrum also reveals two doublets and two singlets that correspond to 3a and 3b, as well as to free ligands 2a and 2b respectively. Based on this NMR experiment, we anticipate that two metallo-ligands are coordinated to one RhI-center to form 3a–c which are in a dynamic equilibrium in solution (Scheme 2).
 | ||
| Fig. 1 Molecular structure of 3d in the crystal. Displacement ellipsoids are shown at the 50% probability level. Counter anion ([BArF4]−) is omitted for clarity. The pendant groups of the metallo-ligands are shown in wire form for clarity. Selected bond lengths (Å) and angles (°): selected bond lengths (Å) and angles (°): P1–Rh1: 2.3072(17); P2–Rh1: 2.2826(17); P2–Rh1–P1: 88.67(6). | ||
 | ||
| Scheme 2 Synthesis of RhI complexes 3a–c and Rh0-complexes 4a–c. | ||
When reaction mixtures containing 3a–c were left standing, orange crystals formed after several weeks, along with an insoluble precipitate. Much to our surprise, the 31P{1H} NMR spectra of the crystals in CD2Cl2 show a complex multiplet resonance at around δ(ppm) = 172.0 (Fig. 2b).
 | ||
| Fig. 2 Simulated (a, AA′XX′) and measured (b) 31P{1H} NMR spectrum of 4a. | ||
The spectra can be simulated as an AA′XX′ spin system, arising from P–P′, Rh–P and Rh–Rh′ interactions (Fig. 2a). From the 1H–103Rh HMQC NMR spectra the chemical shifts of the Rh atoms were identified at around δ(ppm) = −9245 (Fig. S43, ESI‡). The NMR spectroscopic studies therefore indicate that a symmetrical dimeric P2Rh2 species should be present in solution. Single crystals suitable for X-ray diffraction were obtained for all three new species 4a–c and the molecular structure of 4b is depicted in Fig. 3.
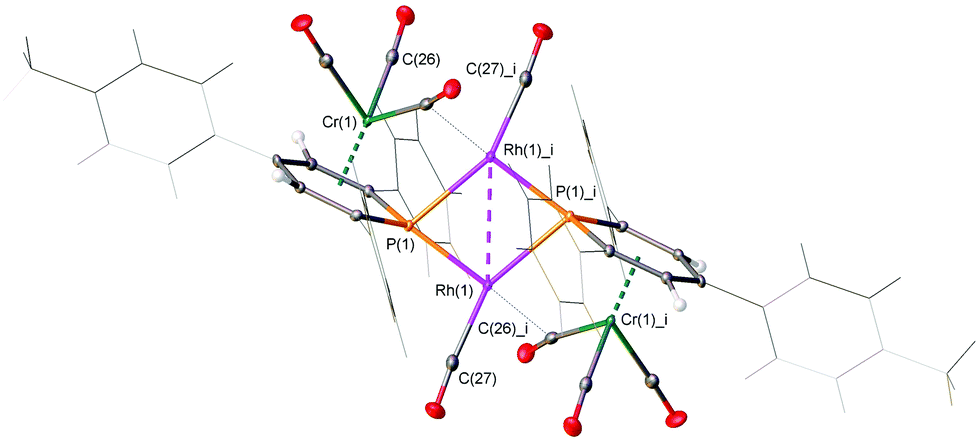 | ||
| Fig. 3 Molecular structure of 4b in the crystal. Displacement ellipsoids are shown at the 50% probability level. The pendant aryl-groups of the metallo-ligands are shown in wire form for clarity. Selected bond lengths (Å) and angles (°): P(1)–Rh(1): 2.1947(6); P(1)–Rh(1):_i: 2.3695(6); Rh(1)–Rh(1)_i: 2.8675(4); P(1)–Cr(1): 2.5214(7). Rh(1)–P(1)–Rh(1)_i: 77.743(18); P(1)–Rh(1)–P(1)_i: 102.260(19). | ||
The crystallographic characterization of 4b reveals the missing η6-PC5, μ2-P coordination mode, in which the phosphinine-π-complex acts as a bridging 2e− ligand and forms a fully planar dimeric P2Rh2 species with Ci geometry and an anti-arrangement of the two phosphinine-Cr(CO)3 metallo-ligands (Chart 1). The P(1)–Rh(1)–P(1)_1 and Rh(1)–P(1)–Rh(1)_i angles are 102.260(19)° and 77.743(18)°, respectively. The distance between the two Rh-atoms (2.8675(4) Å) indicates an interaction between the two Rh0 centers, which is in line with the presence of an overall diamagnetic species (Fig. 2 and vide infra).10,11 One CO-ligand of each Cr(CO)3-fragment is semi-bridging and shows short contacts for C(26)–Rh(1)_i and C(26)_i–Rh(1). The crystallographic characterization of 4a and 4c show similar results (Fig. S50 and S52, ESI‡). However, the most striking feature of 4a–c is the fact that two Rh0 centres are obtained, even though the rhodium atom in the precursor used was in the +1 oxidation state.
To explain the formation of 4a–c, a redox reaction between the Cr(0)- and Rh(I)-species within 3a–c has to be presumed. In fact, it is known that arene–Cr(CO)3 complexes undergo photooxidation under UV-light and decompose in solution to give benzene, CO and a grey-green solid. It has further been shown, for instance, that the photolysis of (η6-C6Et6)Cr(CO)3 leads to photo-fragmentation prior to one-electron oxidation.12 As we also observed decomposition of the π-complex 2a under UV-light (Fig. S4, ESI‡), we anticipated that the formation of 4a–c from 3a–c might also be accelerated by light (Scheme 2). Gratifyingly, the photolysis of a solution of 3a–c in CD2Cl2 with UV-light (λ = 365 nm) leads within 2 h to 4a–c, an insoluble solid and a signal for the η1-phosphinine-RhI complex in the 31P{1H} NMR spectrum (Fig. S4 and S47, ESI‡). The partial decomposition of 3a–c might facilitate the necessary redox reaction and the acquisition of an equivalent of carbon monoxide. Indeed, a survey of the precipitate by XPS analysis reveals the presence of chromium in the oxidation state +3 (Fig. S48, ESI‡). Moreover, the UV-vis spectra of the products consist of intense bands at λmax = 260 nm and less intense broad bands at λmax = 350 nm (Fig. S35, S39 and S44, ESI‡), which can be attributed to MLCT transitions (Fig. S58–S61, ESI‡), confirmed by TDDFT studies. The desired products were obtained in reproducible yields of up to 30%. A similar process has also been observed for bis-η1-mono-phosphaferrocene palladium complexes.13 Similarly, compounds 4a–c can only be formed in a maximum theoretical yield of 50%. Furthermore, the solid state structures of 4a–c are perfectly in line with the 31P{1H} NMR spectra (Fig. 2) of a diamagnetic species in solution. Preliminary CV studies of 4c (Fig. S45 and S46, ESI‡) show that the complex undergoes an irreversible reduction at Ered = −1.59 V (100 mV s−1, vs. Fc*+/0) upon scanning cathodically in THF. The return oxidation wave appears at Eox = −0.78 V. The cyclic voltammogram of 2c shows the same processes but with a shift to the cathodic region (Fig. S45, ESI,‡Ered = −1.98 V; Eox = −0.99 V), due to the energetically destabilized LUMO compared to 4c. Similarities in the shapes of the cyclic voltammograms of 2c and 4c (Fig. S45, ESI‡) suggest that only the phosphinine-Cr(CO)3-fragment in complex 4c was electrochemically active. No other processes were observed upon scanning anodically.
In order to investigate the bonding situation in the Rh-dimer, DFT studies at the ωB97X-D3/def2-TZVP14 level were performed on the truncated model compounds 2a′ and 4a′ (Ar1/Ar2 substituted by H) and on the non-truncated molecule of 4a. The σ-character of the different P–Rh bonds in 4a′ is illustrated by the HOMO−1 and HOMO−4 (Table S5, ESI‡) as well as by an intrinsic bond orbital (IBO) (Table S2, ESI‡, IBO1a/b, IBO2a/b) and a natural bond orbital (NBO) analysis (Table S3, ESI‡).15,16 The IBO in Fig. 4a seems to arise from the P1–Rh1 coordination of the lone pair at the phosphorus atom of the phosphinine–Cr(CO)3 complex 2a′ (Table S2, ESI‡). Fig. 4b supports the suggested Rh–Rh interaction, which are also apparent in the HOMO−8, HOMO−13 and HOMO−14 (Table S5, ESI‡). The two significantly distinct P–Rh bond lengths in the rhomboidal Rh2P2 core can be rationalized by the additional π-character of the shortened P1–Rh1 bond. The shape of the IBO in Fig. 4c already indicates the presence of back donation from Rh-centered d-orbitals to the phosphorus center. The analysis of the bonding in 4a′ by means of NBO-DFT calculations revealed that the Rh-center donates electron density into two equivalent σ*(P–C) orbitals of the phosphinine ring (Table S4a, ESI‡). The energetic stabilization by the donor–acceptor interactions is ΣE(2) = 14.22 kcal mol−1 and is visualized in Fig. 4d for the donation into the σ*(P(1)–C(3)) orbital. The shape of LUMO+6 of 2a′ seems ideal to act as an acceptor orbital for these interactions (Table S4b, ESI‡). The orbital energy in comparison to the LUMO is arguably high (ELUMO = 0.436 eV; ELUMO+6 = 2.472 eV; ΔE((LUMO+6)−LUMO) = 2.036 eV), however the σ*(P–C) orbitals contribute 19.9% to the overall orbital composition of the LUMO+6 and justify the assignment. Stabilizing interactions with antibonding P–C-orbitals of phosphinines and their influence on bond lengths have been previously described by Le Floch and by us.17
 | ||
| Fig. 4 IBOs of 4a′ involving (a) a σ-bond between the P1 and the Rh1 atom, (b) Rh–Rh interactions and (c) back donation between P1–Rh1 (IBOs encompass 70% of the density of the orbital electrons); (d) NBO plots of a donor–acceptor interaction between a σ*(P–C) orbital and Rh-centered d-orbital (isosurface value at ±0.08 a.u.); (e) AIM (Bader) analysis (bond critical points: brown, ring critical points: orange) with Laplacian of the electron density plotted in plane of the Rh2P2 ring. | ||
A Wiberg bond index (WBI) analysis18 for 4a′ provided values of 0.63 and 0.31 for the P1–Rh1 and P1–Rh1′ bonds, respectively, and a WBI of 0.11 for the Rh–Rh interaction. Almost identical values were found for 4a. The AIM analysis19 (Fig. 4e and Fig. S54, S55, ESI‡) shows no BCP for the Rh–Rh interaction, but instead a ring critical point (RCP) at the ring center of 4a and 4a′. In summary, the calculations suggest that the Rh–Rh interaction is rather weak. The topological properties at BCPs and RCPs for selected bonds and rings in 4a and 4a′ are summarized in Table S7 (ESI‡) and complete molecular graphs are shown in Fig. S56 and S57 (ESI‡).
The values of ρBCP for the P1–Rh1 and P1′–Rh1′ bonds in 4a are at the low end for shared interactions but significantly larger than those associated with a closed-shell interaction.20 The smaller values of ρBCP found for the longer P1–Rh1′ and P1′–Rh1 bonds reflect the rhomboidal geometry of the dimer. The negative energy densities HBCP of the ring bonds are typical for covalent contributions.21 The full molecular graph of 4a reveals additional stabilizing closed-shell intramolecular interactions (shown as dashed lines) for i.a. CH···O and Rh···HC, as well as a BCP between Rh1′ and C25 of the carbonyl group that bridges to the Cr(CO)3-fragment (vide supra). Notably several other occupied molecular orbitals (HOMO−1, HOMO−4, HOMO−8, HOMO−12, Table S5, ESI‡) and IBOs suggest interactions between Cr1 and Rh1′ (and Cr1′ and Rh1). The WBI analysis on the other hand gave only values of 0.10 for the Rh–Cr interaction and no bond critical points.
In summary, we have synthesized a complex containing the previously unknown η6-PC5, μ2-P coordination mode of a phosphinine, in which the aromatic phosphorus heterocycle acts both as a 6e−-π-ligand towards a Cr(CO)3-fragment and, additionally, as a bridging 2e− ligand towards a rather rare Rh02(CO)2-core. These diamagnetic coordination compounds were characterized both by NMR spectroscopy and X-ray diffractometry. DFT calculations gave insight into the bonding situation within the phosphinine-bridged Rh2-dimer.
E. Y. and C. M. are grateful for financial support provided by the Sino-German (CSC-DAAD) Postdoc Scholarship Program (57251553). The authors thank the Scientific Computing Service of Freie Universität Berlin for the use of high-performance computing resources as well as M. W. G. M. Verhoeven (Eindhoven University of Technology, The Netherlands) and Dr. G. J. A. Mannie (SPECS Surface Nano Analysis GmbH, Berlin) for their kind support and help with the XPS analysis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. Le Floch and F. Mathey, Coord. Chem. Rev., 1998, 178-180, 771 CrossRef CAS; (b) N. Mézailles, F. Mathey and P. Le Floch, Prog. Inorg. Chem., 2001, 455 Search PubMed; (c) P. Le Floch, Coord. Chem. Rev., 2006, 250, 627 CrossRef CAS; (d) N. T. Coles, A. S. Abels, J. Leitl, R. Wolf, H. Grützmacher and C. Müller, Coord. Chem. Rev., 2021, 432, 213729 CrossRef.
- (a) C. Müller and D. Vogt, Dalton Trans., 2007, 5505 RSC; (b) P. Roesch, J. Nitsch, M. Lutz, J. Wiecko, A. Steffen and C. Müller, Inorg. Chem., 2014, 53, 9855 CrossRef CAS PubMed.
- P. D. Burrow, A. J. Ashe III, D. J. Bellville and K. D. Jordan, J. Am. Chem. Soc., 1982, 104, 425 CrossRef CAS.
- Y. Hou, Z. Li, Y. Li, P. Liu, C. Y. Su, F. Puschmann and H. Grützmacher, Chem. Sci., 2019, 10, 3168 RSC.
- S. Giese, K. Klimov, A. Mikehászi, Z. Kelemen, D. S. Frost, S. Steinhauer, P. Müller, L. Nyulászi and C. Müller, Angew. Chem., Int. Ed., 2021, 60, 3581 CrossRef CAS PubMed.
- M. Doux, L. Ricard, F. Mathey, P. Le Floch and N. Mézailles, Eur. J. Inorg. Chem., 2003, 687 CrossRef CAS.
- P. A. Cleaves and S. M. Mansell, Organometallics, 2019, 38, 1595 CrossRef CAS.
- C. Elschenbroich, J. Six and K. Harms, Chem. Commun., 2006, 3429 RSC.
- (a) J. Deberitz and H. Nöth, Chem. Ber., 1970, 103, 2541 CrossRef CAS; (b) H. Vahrenkamp and H. Nöth, Chem. Ber., 1972, 105, 1148 CrossRef CAS.
- For paramagnetic Rh0 complexes see for example: B. de Bruin and D. G. H. Hetterscheid, Eur. J. Inorg. Chem., 2007, 211 CrossRef CAS.
- A. F. Heyduk, A. M. Macintosh and D. G. Nocera, J. Am. Chem. Soc., 1999, 121, 5023 CrossRef CAS.
- (a) A. Gilbert and J. M. Kelly, Z. Naturforsch., 1976, 31b, 1091 CrossRef CAS; (b) R. G. Compton, R. Barghout, J. C. Eklund, A. C. Fisher, S. G. Davies, M. R. Metzler, A. M. Bond, R. Colton and J. N. Walter, J. Chem. Soc., Dalton Trans., 1993, 3641 RSC.
- X. Sava, L. Ricard, F. Mathey and P. Le Floch, Chem. – Eur. J., 2001, 7, 3159 CrossRef CAS.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; (c) J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834 CrossRef CAS PubMed.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211 CrossRef CAS.
- (a) A. Moores, T. Cantat, L. Ricard, N. Mézailles and P. Le Floch, New J. Chem., 2007, 31, 1493 RSC; (b) F. Wossidlo, D. S. Frost, J. Lin, N. T. Coles, K. Klimov, M. Weber, T. Böttcher and C. Müller, Chem. – Eur. J., 2021, 27, 12788 CrossRef CAS PubMed.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083 CrossRef CAS.
- R. F. W. Bader and Acc Chem, Res., 1985, 18, 9 CAS.
- (a) P. Macchi and A. Sironi, Coord. Chem. Rev., 2003, 238-239, 383 CrossRef CAS; (b) E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529 CrossRef CAS.
- (a) D. Cremer and E. Kraka, Angew. Chem., Int. Ed., 1984, 23, 627 CrossRef; (b) F. Cortés-Guzmán and R. Bader, Coord. Chem. Rev., 2005, 249, 633 CrossRef.
Footnotes |
| † Dedicated to Prof. Christian Bruneau. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2113811–2113814. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc01817a |
| § These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |