
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sigma profiles in deep learning: towards a universal molecular descriptor†
Dinis O.
Abranches
 ,
Yong
Zhang
,
Edward J.
Maginn
and
Yamil J.
Colón
*
,
Yong
Zhang
,
Edward J.
Maginn
and
Yamil J.
Colón
*
Department of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: ycolon@nd.edu
First published on 11th April 2022
Abstract
This work showcases the remarkable ability of sigma profiles to function as molecular descriptors in deep learning. The sigma profiles of 1432 compounds are used to train convolutional neural networks that accurately correlate and predict a wide range of physicochemical properties. The architectures developed are then exploited to include temperature as an additional feature.
In the field of machine learning (ML), a deep neural network (DNN) is a mathematical model that functions as a universal approximator, being able to capture and reproduce highly complex relationships between independent (features) and dependent (labels) variables.1,2 Put simply, a DNN is a collection of layers of fully-connected nodes. The first layer of the DNN is the input layer (features) while the last layer contains the output (label). Each node in a hidden layer represents a linear combination whose inputs are the weighted outputs of all nodes in the previous layer. The linear combination is then passed through an activation function, which adds non-linearity to the network, and fed into the nodes of the following layer.
The ability of DNNs to correlate variables whose relationship is unknown or too complex to be derived is attracting a great deal of interest in chemistry-related fields,3–9 namely in the prediction of physicochemical properties,6 chemical synthesis,7 and drug design.8,9 Common to these studies is the necessity to represent molecules in a way that can be used as an input to DNNs. In other words, because the input of a DNN is a set of numerical values, molecular structures must be converted into a set of features that describes their intrinsic chemical nature (i.e., atom types, connectivity, polarity, etc.).
Some of the molecular representations proposed include string-based vectors such as SMILES or SELFIES,10,11 molecular fingerprints,12 molecular graphs,13 and Coulomb matrixes.14 Despite the achievements obtained using these molecular representations, they present several shortcomings.5,15 For instance, they encapsulate little chemical information beyond atom type and connectivity. Polarity and the potential for non-covalent interactions are missing. Furthermore, the size of these representations depends on the size of the molecule: the larger the molecule, the larger the vector or matrix used to represent it. Thus, the input size of a DNN must be made as large as the largest molecule available in the dataset of interest. This leads to the development of complex neural networks (NNs) that possess a large number of trainable parameters and, thus, need very large datasets to be properly fitted. Finally, these representations are abstract collections of numerical values that cannot be readily understood by humans. The point can be made that having abstract molecular representations hinders the understanding of how DNN models work and how they can be improved.
Although it is yet unclear whether a universal representation of molecular structures exists,5,15 the present work proposes a new molecular descriptor for deep learning. Aiming at mitigating the disadvantages of classical methodologies discussed above, the concept of the sigma profile (σ-profile) is here proposed as a promising candidate for a universal molecular descriptor. Initially developed by Klamt to be used in the COSMO-RS thermodynamics model,16,17 σ-profiles are unnormalized histograms of the screened charge of molecules. They quantify the polarity (or lack thereof) of a molecule. They are obtained by optimizing the geometry of molecules embedded in the COnductor like Screening MOdel (COSMO) continuum solvation model, using density-functional theory (DFT). The surface area of the molecule is divided into segments and the screened charge (σ) is calculated for each segment. Then, a histogram, P(σ), is built that reflects the probability distribution of σ. The product between P(σ) and the total area of the molecule gives rise to the σ-profile. A more detailed description and examples are provided in Section S1 of the ESI.†
Sigma profiles provide several advantages over classical molecular representations in deep learning. First, they are a holistic representation of molecules; rather than focusing on atom types and their connectivity, σ-profiles represent molecules as surfaces composed of charged segments. Given their strong quantum chemistry foundation, they capture subtle effects such as polarizability and electron density asymmetry across covalent bonds between atoms with different electronegativities (Fig. S1, ESI†). As such, they are particularly well suited to capture and describe non-covalent interactions (and properties that depend strongly on them) and are intuitively easy to understand. Another advantage of σ-profiles is their ability to describe molecules of any size (Fig. S2, ESI†). Because they are unnormalized histograms of screened charge area, the size of the molecule does not change the σ range of the σ-profile, thus, not changing the size of the DNN input.
It is worth mentioning that σ-profiles have been successfully used in the past, outside of the COSMO-RS framework, in methodologies ranging from the well-known quantitative structure–activity relationship (QSAR) correlations18 to ML approaches.19,20 However, these works do not use the entire σ-profile as a molecular descriptor, relying instead on a small subset of the σ-profile (or quantities calculated from it) together with other, unrelated molecular descriptors.
The main objective of this Communication is to demonstrate that the σ-profile can function on its own as a universal molecular descriptor in deep learning. To do so, the σ-profile database developed by Mullins et al.21 was used to develop convolutional neural networks (CNNs) that fit and predict six separate physicochemical properties: molar mass, normal boiling temperature, vapor pressure at 25 °C, density at 20 °C, refractive index at 20 °C, and aqueous solubility at 25 °C. The size of these datasets ranged from 1432 for the most data rich property (molar mass) to 327 for the most data scarce property (aqueous solubility). Then, the CNN architectures developed were exploited such that thermodynamic conditions (temperature) could be used as an additional feature to fit and predict temperature-dependent properties available in the original database used, namely density (−200 °C to 240 °C), refractive index (−100 °C to 134 °C), and aqueous solubility (−3 °C to 40 °C). The datasets and Python code used throughout this work are available in a GitHub repository.‡
The σ-profiles used in this work were taken from the freely available Mullins et al.21 database (Section S1, ESI†). This database was chosen due to its previous extensive use and validation within the framework of COSMO-RS and large chemical diversity. Each σ-profile contained in the Mullins et al.21 database is composed of 51 points (σ, P(σ)·A). The σ values are comprised within the range −0.025 e Å−2 and 0.025 e Å−2, in intervals of 0.001 e Å−2. Thus, the 51 P(σ) A values of each molecule were used, in the form of vectors of size (51, 1), as the input for the CNNs developed in this work (Fig. S3, ESI†). All physicochemical properties studied in this work were taken, when available, from the CRC Handbook of Chemistry and Physics (Internet version).22 Dataset details, their distributions, and normalization procedures are provided in the ESI.†
The general architecture for the DNNs developed in this work is based on convolution layers, as depicted in Fig. 1. CNNs are a powerful subset of DNNs and are especially suited for highly correlated features such as temporal data or the σ-profiles here explored.23 Each CNN developed in this work possesses between one and two convolution layers, one and two pooling layers, and one or two dense layers. The number of filters, kernel size, and number of strides of each convolution layer, the type and size of each pooling layer, the number of nodes of each dense layer, and the activation function of all nodes of the NNs were treated as hyperparameters to be tuned.
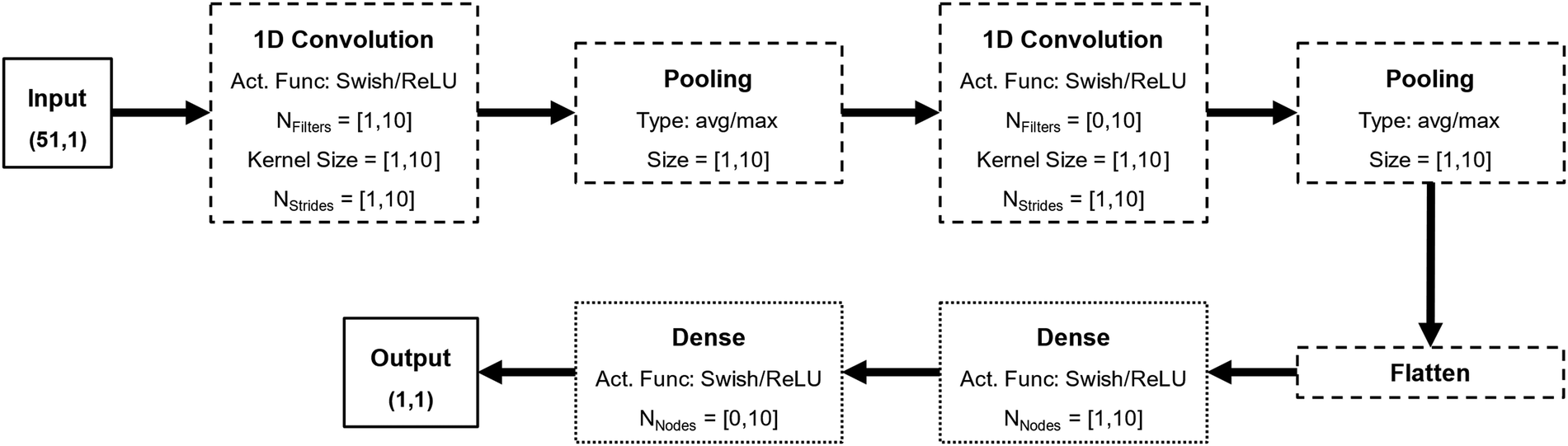 | ||
| Fig. 1 Generic architecture of the convolutional neural networks developed in this work, including the main hyperparameters and highlighting the input and output of the network (full lines), the convolution module (dashed lines), and the fully connected (dense) module (dotted lines). | ||
The computational details of the hyperparameter tuning and NN fitting are discussed at length in Section S3 of the ESI.† Briefly, the hyperparameter tuning of the architecture of each CNN was performed in two stages using the open source software package Sherpa.24 First, Bayesian optimization was performed, to converge to an initial guess for the architecture hyperparameters. Then, local search algorithms were used to tune both the architecture hyperparameters and fitting hyperparameters. Each CNN was fitted using the Adam optimizer with early-stopping. To prevent overfitting and maximize the generalization capability of the networks, each data set (σ-profile and property of interest) was split into fitting and testing sets (90/10). Then, both during the hyperparameter tuning and fitting of the final architecture of the network, each fitting dataset was further split into training and validation sets (80/20). This split was done at random, using stratified splitting, at each repetition of each trial. Finally, the number of fitting (trainable) parameters of each NN was constrained to be lower than one third of the total number of data points of each data set (Section S3, ESI†). Note that the testing sets were never used during the hyperparameter tuning or fitting stages, being only used to independently evaluate the final performance of each CNN.
Using the procedures summarized above and described at length in Section S3 of the ESI,† six CNNs were developed to fit and predict molar mass, normal boiling temperature, vapor pressure at 25 °C, density at 20 °C, refractive index at 20 °C, and aqueous solubility at 25 °C. The performance of the CNNs for molar mass, normal boiling temperature, and vapor pressure at 25 °C are depicted in Fig. 2, while the performance for the remaining datasets (density, refractive index, and aqueous solubility) are reported in Section S4.1 (ESI†). Note that these last three datasets are not the focus of the work at this point, as they will be explored later to show that temperature can be an additional input in the modular architectures developed.
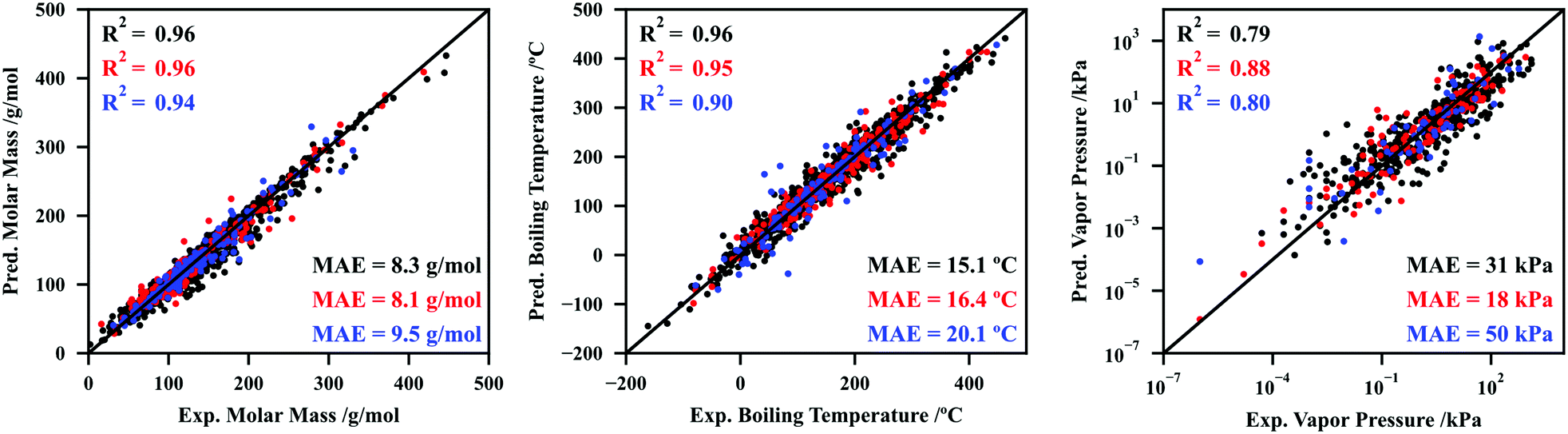 | ||
| Fig. 2 Performance (predicted property vs. experimental property) of the convolutional neural networks developed in this work for molar mass (left), normal boiling temperature (middle), and vapor pressure at 25 °C (right). Black, red, and blue colors represent the results for the training, validation, and testing sets, respectively. The coefficient of determination (R2) and mean absolute error (MAE) are also included. | ||
Fig. 2 and Fig. S14 (ESI†) reveal an overall excellent performance of the CNNs here developed. Regarding the testing sets, coefficients of determination of 0.94, 0.90, and 0.80 were attained for molar mass, boiling temperature, and vapor pressure, respectively. This is quite remarkable, especially considering the relatively small size of these networks and datasets, and the complex underlying relationships that exist between the molecular descriptor (σ-profile) and each property. For instance, although the prediction of molar masses may appear trivial, note that the atom type or atomic weight is not explicitly hardcoded in σ-profiles. The prediction is made no simpler for the remaining properties, as the CNN must learn the patterns between σ-profile and material structure and non-covalent interactions.
Following the results reported in Fig. 2 and Fig. S14 (ESI†), it becomes apparent why σ-profiles are being claimed in this work as universal molecular descriptors. Using CNNs with relatively few trainable parameters, σ-profiles perform remarkably well for a wide variety of organic and inorganic compounds, and a wide range of physicochemical properties.
Having demonstrated the accuracy of the deep learning methodologies developed in this work, the modular architecture concept is now explored. As highlighted in Fig. 1, the architecture of the NNs developed in this work can be seen as the junction of a convolutional module and a dense model. In fact, the main objective of the convolution module is to transform the initial σ-profile input into a smaller, abstract feature set that can be fed into the dense module. Given this, it is reasonable to speculate that further information can be fed into the network by adding inputs (features) between the convolution and dense modules. This new information can be, for instance, the thermodynamic conditions of the system, such as temperature (Fig. 3).
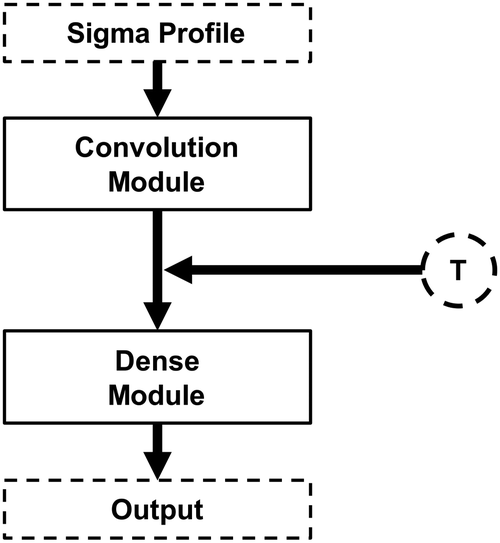 | ||
| Fig. 3 Schematic illustration of the convolutional neural networks explored in this work using both the σ-profile and temperature as features. | ||
To test the hypothesis presented in the previous paragraph, CNNs were developed to correlate and predict the temperature-dependent density, refractive index, and aqueous solubility. The hyperparameter tuning and intermediate results are reported in Section S4.2 (ESI†). The performances of the three CNNs developed are depicted in Fig. 4.
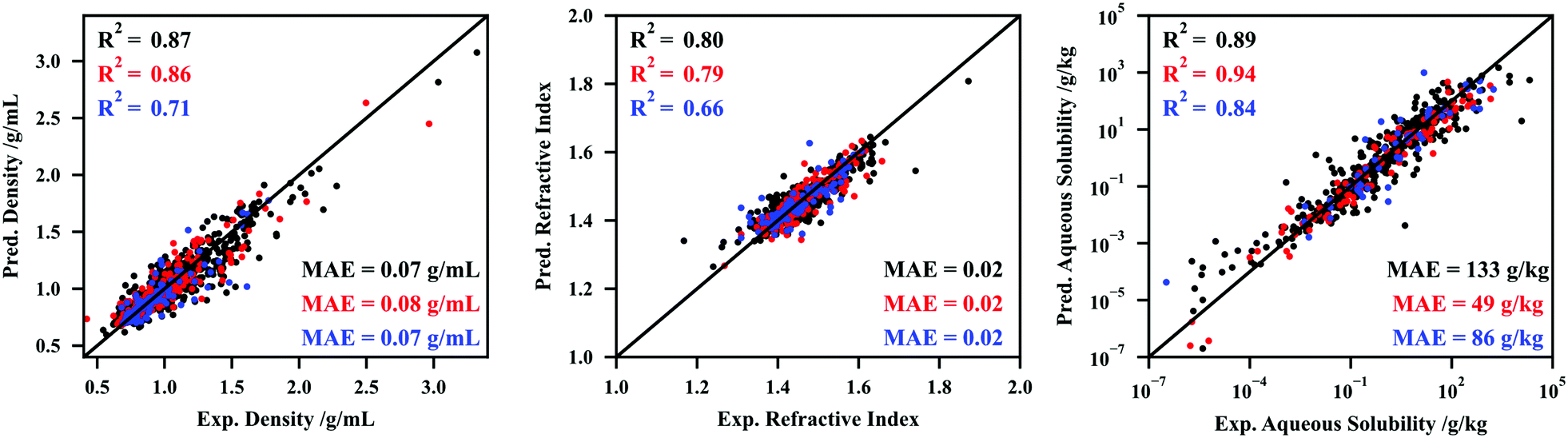 | ||
| Fig. 4 Performance (predicted property vs. experimental property) of the convolutional neural networks developed in this work for the temperature-dependant properties density (left), refractive index (middle), and aqueous solubility (right). Black, red, and blue colors represent the results for the training, validation, and testing sets, respectively. The coefficient of determination (R2) and mean absolute error (MAE) are also included. | ||
Fig. 4 shows that the deep learning methodologies developed in this work can be made more flexible by adding thermodynamic inputs in the junction between the convolution and densely connected modules of the networks. The density and refractive indexes are available for both liquids and solids, and no distinction is made between the two phases in the input to the CNNs, which adds an additional layer of complexity to the process of predicting these datasets. The performance for aqueous solubility is particularly robust. Note that the higher performance of this CNN in the validation set is related to the randomness associated with the final fitting (performing several fittings, attempting to maximize the performance in the validation set).
To conclude, this work showcases, for the first time, the ability of σ-profiles to function as universal molecular descriptors in deep learning. The CNNs here developed were able to accurately fit and predict several different physicochemical properties, and it was shown that thermodynamic conditions can also be used as additional inputs to broaden the applicability of the models. Among all other advantages mentioned, this work shows that σ-profiles can extend the use of deep learning methodologies to areas where datasets are relatively small and scarce. The bridge between small datasets and deep learning, made possible using σ-profiles, will be explored in the future for more complex properties and molecules. Improving the level of theory used to obtain σ-profiles, namely basis sets, DFT functionals, and grid sizes is expected to have an impact on this deep learning framework, which will also be explored in future work. On the other hand, although the necessary quantum chemistry calculations to obtain the σ-profile of a simple organic molecule can be performed in a matter of seconds (or minutes, for large molecules) on most modern workstations, this step can still be viewed as a computational bottleneck of the framework developed here. This may be mitigated by building extensive σ-profiles databases in the open literature or developing semi-empirical methodologies to estimate them.
This work was supported by the U.S. Department of Energy via subcontract 630340 from Los Alamos National Laboratory. The authors acknowledge the Center for Research Computing (CRC) at the University of Notre Dame for providing computational resources.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. LeCun, Y. Bengio and G. Hinton, Nature, 2015, 521, 436–444 CrossRef CAS PubMed.
- J. Schmidhuber, Neural Networks, 2015, 61, 85–117 CrossRef PubMed.
- G. B. Goh, N. O. Hodas and A. Vishnu, J. Comput. Chem., 2017, 38, 1291–1307 CrossRef CAS PubMed.
- E. N. Muratov, J. Bajorath, R. P. Sheridan, I. V. Tetko, D. Filimonov, V. Poroikov, T. I. Oprea, I. I. Baskin, A. Varnek, A. Roitberg, O. Isayev, S. Curtalolo, D. Fourches, Y. Cohen, A. Aspuru-Guzik, D. A. Winkler, D. Agrafiotis, A. Cherkasov and A. Tropsha, Chem. Soc. Rev., 2020, 49, 3525–3564 RSC.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360–365 CrossRef CAS PubMed.
- Q. Zang, K. Mansouri, A. J. Williams, R. S. Judson, D. G. Allen, W. M. Casey and N. C. Kleinstreuer, J. Chem. Inf. Model., 2017, 57, 36–49 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, F. Sandfort, M. H. S. Segler and F. Glorius, Chem. Soc. Rev., 2020, 49, 6154–6168 RSC.
- Y. Jing, Y. Bian, Z. Hu, L. Wang and X.-Q. S. Xie, AAPS J., 2018, 20, 58 CrossRef PubMed.
- L. Zhang, J. Tan, D. Han and H. Zhu, Drug Discovery Today, 2017, 22, 1680–1685 CrossRef PubMed.
- M. Hirohara, Y. Saito, Y. Koda, K. Sato and Y. Sakakibara, BMC Bioinf., 2018, 19, 526 CrossRef CAS PubMed.
- M. Krenn, F. Häse, A. Nigam, P. Friederich and A. Aspuru-Guzik, Mach. Learn. Sci. Technol., 2020, 1, 045024 CrossRef.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed.
- S. Kearnes, K. McCloskey, M. Berndl, V. Pande and P. Riley, J. Comput.-Aided Mol. Des., 2016, 30, 595–608 CrossRef CAS PubMed.
- M. Rupp, A. Tkatchenko, K.-R. Müller and O. A. von Lilienfeld, Phys. Rev. Lett., 2012, 108, 058301 CrossRef PubMed.
- O. A. von Lilienfeld, Int. J. Quantum Chem., 2013, 113, 1676–1689 CrossRef CAS.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- A. Klamt, COSMO-RS: from quantum chemistry to fluid phase thermodynamics and drug design, Elsevier, 2005 Search PubMed.
- G. Járvás, C. Quellet and A. Dallos, Fluid Phase Equilib., 2011, 309, 8–14 CrossRef.
- O. Nordness, P. Kelkar, Y. Lyu, M. Baldea, M. A. Stadtherr and J. F. Brennecke, J. Mol. Liq., 2021, 334, 116019 CrossRef CAS.
- H. Benimam, C. S. Moussa, M. Hentabli, S. Hanini and M. Laidi, J. Chem. Eng. Data, 2020, 65, 3161–3172 CrossRef CAS.
- E. Mullins, R. Oldland, Y. A. Liu, S. Wang, S. I. Sandler, C.-C. Chen, M. Zwolak and K. C. Seavey, Ind. Eng. Chem. Res., 2006, 45, 4389–4415 CrossRef CAS.
- J. R. Rumble, CRC Handbook of Chemistry and Physics, CRC Press/Taylor & Francis, Boca Raton, United States, 102nd edn, 2021 Search PubMed.
- S. Albawi, T. A. Mohammed and S. Al-Zawi, 2017 International Conference on Engineering and Technology (ICET), IEEE, 2017, pp. 1–6 Search PubMed.
- L. Hertel, J. Collado, P. Sadowski, J. Ott and P. Baldi, SoftwareX, 2020, 12, 100591 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc01549h |
| ‡ GitHub repository: https://github.com/MaginnGroup/SP_ML_CC |
| This journal is © The Royal Society of Chemistry 2022 |