
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient covalent capping of carbon and gold with TEMPO for catalysis and spin writing†‡
Vitalijs
Romanovs
*a,
Valery
Sidorkin
 *b,
Evgeniya
Doronina
b and
Viatcheslav
Jouikov
*c
*b,
Evgeniya
Doronina
b and
Viatcheslav
Jouikov
*c
aLatvian Institute of Organic Synthesis, LV-1006 Riga, Latvia. E-mail: vitalijs.romanovs@inbox.lv
bA. E. Favorsky Irkutsk Institute of Chemistry, 664033 Irkutsk, Russia. E-mail: svf@irioch.irk.ru
cUMR 6226 ISCR, University of Rennes 1, 35042 Rennes, France. E-mail: vjouikov@univ-rennes1.fr
First published on 31st August 2022
Abstract
Covalent immobilization of TEMPO at carbon and gold via an aliphatic –(CH2)6– linker was achieved via cathodic grafting of a diamagnetic precursor, tetramethylpiperidine, with subsequent >NH to >NO oxidation to give TEMPO-capped paramagnetic interfaces (ΓTEMPO = 5.2 × 10−10 mol cm−2); catalytic and spin switching potential of the thus prepared systems was demonstrated.
Immobilization of 2,2,6,6-tetramethyl piperidyl oxide (TEMPO) stable radicals at solid conducting supports is a key element in a large number of applications1–3 related to interfaces with permanent magnetic character that are sought for sensing,4 electronic,5,6 energy storage,6–11 catalysis,12–18 biomedical,11,19 and other fields. Two critical issues are to be dealt with here: (i) simple and reliable immobilization/fixation of TEMPO at the support and (ii) a long, chemically stable and atom-economical spacer attaching TEMPO to the support.
Covalent grafting of TEMPO is obviously more advantageous than its mechanical incorporation into composites because of its much smaller consumption, reducing the thickness of the immobilized phase to a monolayer, avoiding its leakage from the interface and providing better solvent and mechanical stability.5,8,16 While diamagnetic substrates can be easily grafted via their diazonium derivatives,20 the radical character of TEMPO excludes this radical-based process.5 However, the cationic nitroxonium form of TEMPO obtained through its disproportionation in an acid medium can be grafted from its diazonium derivative and subsequently reduced back to a nitroxyl radical by cathodic cycling.5
Non-radical covalent immobilizations of TEMPO at carbons and CNTs were also reported,8,21–23 with a common drawback being a specific pre-treatment of the substrate or precursor, while the resulting “scaffold” is either excessively bulky or chemically non-inert.
When a well-shaped EPR response of the grafted radical centers is required for operating their spin states,24 a sufficiently long spacer separating them from the surface is needed to prevent masking their EPR signal by surface electronic conductivity.10,23–26 A relatively long linker is as well required to enable a pseudo-diffusional behavior of the immobilized radical units,27 which is critical for catalytic applications. Due to their chemical inertness, flexibility and smaller footprints on the surface compared to those of TEMPO itself, alkyl linkers appear most advantageous for this purpose.
We report here on a simple and efficient covalent attachment of TEMPO to carbonaceous and gold interfaces using non-diazonium alternative grafting28,29via an aliphatic linker (CH2)n (n = 6) preserving the redox reactivity and distinct EPR response of TEMPO.
An easy to handle diamagnetic precursor of TEMPO fitted with a (CH2)6 linker has been prepared from commercially available 4-hydroxy-2,2,6,6-tetramethyl-piperidine (TMP-OH) and 1-bromo-6-chloro-hexane (ESI†) (Scheme 1), chosen because of its moderately long alkyl chain and availability.
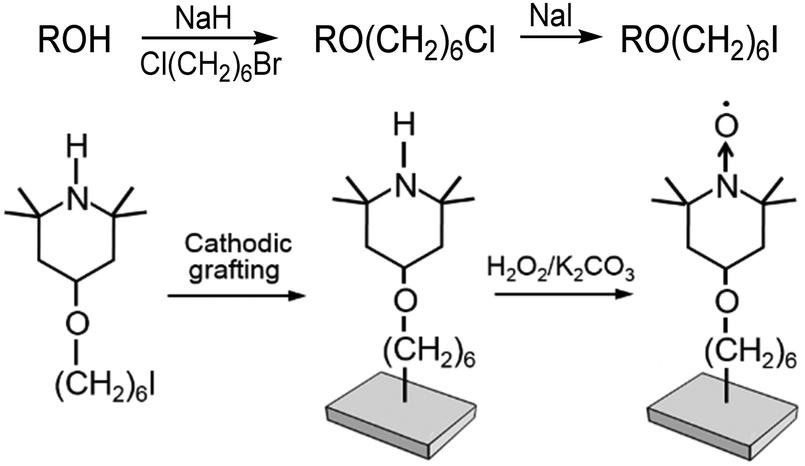 | ||
| Scheme 1 Synthesis (R = 4-TMP), immobilization of TMP-O(CH2)6I and oxidation of the grafted N–H functions to N→O. | ||
Nucleophilic grafting of TMP-O(CH2)6I to carbonaceous supports was realized via an SN2-like process upon their activation29 by scanning between −1.5 and −2.0 V vs. Ag/AgCl (Fig. 1) until total blocking of the electrode by the grafted units.
 | ||
| Fig. 1 Cathodic grafting of TMP-O(CH2)6I onto GC (14 consecutive scans from −1.4 to −2.1 V) and gold (12 scans from 0 to −1.5 V). CH3CN/0.1 M Bu4NPF6, ν = 50 mV s−1. T = 293 K. | ||
Note that the polarity of CH3CN is not high enough to allow a one-electron reduction of the C–I bond of the hexyl linker,30 thus excluding the radical grafting pathway. Various carbonaceous supports were functionalized this way: bulky glassy carbon (GC) and CG cloth, HOPG, natural Ceylon graphite, technical graphite rods and an HB pencil lead.
For a covalent attachment of the TMP precursor to Au, spontaneous in situ formation of oxidative addition intermediate TMP-O(CH2)6–Au(II)-I and its one-electron reduction (Fig. 1) at moderate cathodic potentials (E ≅ −1 V)24,28 were exploited. A similar procedure can be used for Pt (Fig. S1, ESI†), Ag and Cu as well.
The thus prepared GC and Au interfaces both show an irreversible signal (Ep = 1.18 V) of oxidation of TMP units (Fig. 2A). Its integration (Q = 3.62 μC for an electrode of ∅ = 3 mm) provides an estimate of their efficient surface concentration as Γ∝(TMP) ≅ 5.2 × 10−10 mol cm−2. This value is surprisingly close to that found for TEMPO thiol-attached to gold via a (CH2)15C(O)NH linker (4.6 × 10−10 mol cm−2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26) and is two times higher than the coverage of an ideally flat surface at a 90% maximal hexagonal packing (2.7 × 10−10 mol cm−2). Apparent maximal coverage at GC is Γ∝(TMP) ≅ 5.4 × 10−9 mol cm−2, approaching that observed for grafting Fc(CH2)6 onto GC (10 × 10−9 mol cm−230). Since nucleophilic grafting specifically provides a single layer coverage, these high values evidently stem from the roughness of the GC interface.
26) and is two times higher than the coverage of an ideally flat surface at a 90% maximal hexagonal packing (2.7 × 10−10 mol cm−2). Apparent maximal coverage at GC is Γ∝(TMP) ≅ 5.4 × 10−9 mol cm−2, approaching that observed for grafting Fc(CH2)6 onto GC (10 × 10−9 mol cm−230). Since nucleophilic grafting specifically provides a single layer coverage, these high values evidently stem from the roughness of the GC interface.
 | ||
| Fig. 2 Voltammetry of the grafted interfaces. (A) Three anodic cycles at a GC electrode: irreversible oxidation of the grafted TMP units. (B) Same electrode (12 cycles) after converting TMP to TEMPO with H2O2/K2CO3; reversible oxidation of TEMPO. (C) TEMPO-capped HOPG. (D) TEMPO-capped Au. CH3CN/0.1 M Bu4NPF6, ν = 50 mV s−1. T = 293 K. The shadowed zones show current integration. | ||
After oxidizing the grafted TMP units by dipping into the H2O2/K2CO3 solution (ESI†), the interface shows a reversible peak of a TEMPO+/TEMPO redox pair (Eo = 0.69 V) whose coulometry attests to a quantitative TMP to TEMPO conversion (Fig. 2B). In CH3CN/0.1 M Bu4NPF6 solution, the immobilized redox couple TEMPO+/TEMPO is quite stable upon repetitive cycling (Fig. 2B–D); cyclic voltammetry and two-step potentiostatic chronoamperometry (at Ep) show no visible diminishing of the amount of immobilized TEMPO (after > 105 cycles, see Fig. S3, ESI†).
The FTIR reflection spectra of the grafted GC interfaces before and after peroxide oxidation (Fig. 3D) show the disappearance of the characteristic N–H vibration modes (3360, 700 cm−1) and the appearance of the band of N → O (960 cm−1). The SEM images of the grafted interface (Fig. 3A–C) show smoothening of the micro relief of the bare GC surface by the immobilized layers, while EDS atom analysis attests to the presence of C, N and O atoms at the gold interface (Fig. S4, ESI†).
 | ||
| Fig. 3 The SEM images of the GC surface: (A) before grafting, (B) after immobilization of 4-hexyloxy-piperidine and (C) after the oxidation of the grafted TMP to TEMPO. (D) The FTIR reflectance spectra of the GC interface functionalized with (a) TMP and (b) TEMPO. (E) The Nyquist plots for the reduction of chloranil at (a) TMP and (b) TEMPO capped interfaces, E0 = −0.1 V, f = 0.24 MHz to 0.1 Hz. ΔE = 10 mV (see also Fig. S2, ESI†). (F) The EPR spectra of TEMPO covalently grafted via an O(CH2)6 linker to: (a) GC, (b) a bundle of GC fibres, and (c) 0.5 mm HB pencil lead. | ||
The (CH2)6O-TMP-covered GC interface is EPR silent but upon its oxidation with H2O2, a distinct well-resolved triplet (Fig. 3F) of TEMPO appears (g = 2.006, aN = 15 G); similar spectra were obtained with a TEMPO-grafted HOPG stripe and 0.5 mm HB pencil lead. Using a bundle of ca. 30 GC fibers (5.56 μm in diameter) functionalized with (CH2)6O-TEMPO and fixing the static field at the central line of the nitrogen spectrum (H = 3374 G), the word “SPIN” (Fig. 4) was written in Morse code switching between paramagnetic and diamagnetic states of the interface by on-off polarization steps (E = 0.2 V for ON and E = 1.2 V for OFF states). Note that in contrast to the interface with redox-switchable n-propyl-linked silatranyl radicals,24 the paramagnetic state corresponds here to lower voltage, so the spin writing with TEMPO is rather a “spin-erasing” technique with a stationary spin-ON state (for permanent redox-switching of TEMPO/TEMPOL couple, see (Fig. S5, ESI†)).
 | ||
| Fig. 4 Commutation of the EPR response of the TEMPO+/TEMPO redox couple immobilized at GC fibres (∅ 5 μm): the word “SPIN” written in inverted-key Morse code. Spin switching commands: EOFF(“0”) = 1.2 V, EON(“1”) = 0.2 V, static field 3374 Gauss, and modulation amplitude 2 G. T = 293 K. | ||
The thus prepared TEMPO-modified interfaces were probed for catalytic alcohol oxidation, one of the major applications of TEMPO.12–18,31–33 The voltammetric responses of a gold (CH2)6O-TEMPO-capped interface (in CH3CN/0.1 M Bu4NPF6) in the presence of PhCH2OH (0.02 mol L−1) and 2,6-lutidine (0.08 mol L−1) used as a base well show all characteristic features of a catalytic process: increase of the current with respect to the response of the catalyst in the absence of the alcohol (Fig. 5A–C), disappearance of the reverse current of the reversible redox pair of the catalyst; and, seen for sharper shaped voltammograms (Fig. 5B), a backward kinetic shift of the oxidation peak because of fast consumption of the active form of the catalyst (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O+ cation) in a follow up catalytic reaction. The efficiency of recovery of the nitroxyl radical (Fig. 5A) due to high turnover of the catalytic reaction with the alcohol (ipcat ≅ 7.5×ipTEMPO) is close to that of TEMPO thiol-attached to Au nano particles.31 For comparison, under homogeneous conditions (CTEMPO = 7 mmol L−1 in CH3CN), this ipcat/ip ratio was only attained with a 100× excess of PhCH2OH.32,33
O+ cation) in a follow up catalytic reaction. The efficiency of recovery of the nitroxyl radical (Fig. 5A) due to high turnover of the catalytic reaction with the alcohol (ipcat ≅ 7.5×ipTEMPO) is close to that of TEMPO thiol-attached to Au nano particles.31 For comparison, under homogeneous conditions (CTEMPO = 7 mmol L−1 in CH3CN), this ipcat/ip ratio was only attained with a 100× excess of PhCH2OH.32,33
 | ||
| Fig. 5 Catalysis of the oxidation of benzyl alcohol by the immobilized couple TEMPO+/TEMPO. (A) TEMPO-capped Au electrode in CH3CN/0.1 M Bu4NPF6 + 2,6-lutidine (0.08 mol L−1) without (broken line) and in the presence (solid) of PhCH2OH: (1) 5, (2) 10, and (3) 30 mmol L−1, ν = 250 mV s−1. (B and C) TEMPO-capped (B) GC and (C) graphite paste electrodes in H2O/K2CO3 (pH 10) in the presence of PhCH2OH: (1) 2, (2) 4 and (3) (B: 5 mmol L−1, C: 20 mmol L−1), ν = 100 mV s−1. T = 293 K. | ||
In aqueous alkali media (pH 10), the catalytic feature at the TEMPO-functionalized Au electrode is more complex (Fig. S5, ESI†), which is yet to be entirely rationalized (see Fig. S6, ESI†). Under the same conditions, the responses of TEMPO-modified graphitic electrodes (Fig. 5B and C), more suitable for large scale catalytic oxidations,16 show a rapid loss of the reversibility of the NO+/NO˙ system, while the catalytic current increases with the concentration of PhCH2OH, in line with the previous reports.16,31–34 It is to be noted that with an excellent proper stability of the grafted TEMPO layer (see Fig. S3, ESI†), its partial degradation occurs during large scale alcohol oxidation due to undesired side reactions of TEMPO, also reported in other works.16,31
Thus, the cathodic covalent functionalization of various carbon and gold interfaces with TEMPO using a universal aliphatic linker, equally suitable for both supports, was shown to be very advantageous in terms of easily available precursors (including those with different CH2 chain lengths), a very simple experimental protocol and good grafting efficiency. This method provides an excellent stability of TEMPO-functionalized interfaces; with this, an easy and deliberate CH2-incremental variation of the length of the attaching spacer and hence the thickness of the underlying hydrophobic layer is possible, allowing the creation of interfaces with promising properties for spin-commutation, catalysis and other applications. Further work on immobilized radical systems is under progress.
The authors are grateful to European PostDoc Latvia #1.1.1.2/VIAA/3/19/577 (V. R.) and PHC OSMOSE Project no: 48362YK (V. R. and V. J.) for financial support.
Conflicts of interest
The authors have no conflicts to declare.References
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118/9, 4834–4885 CrossRef PubMed.
- Z. Zhou and L. Liu, Curr. Org. Chem., 2014, 18/4, 459–474 CrossRef.
- A. Das and S. S. Stahl, Angew. Chem., Int. Ed., 2017, 56/30, 8892–8897 CrossRef PubMed.
- S. Abdellaoui, K. L. Knoche, K. Lim, D. P. Hickey and S. D. Minteer, J. Electrochem. Soc., 2016, 163/4, H3001–H3005 CrossRef.
- C. Cougnon, S. Boisard, O. Cador, M. Dias, E. Levillain and T. Breton, Chem. Commun., 2013, 49, 4555–4557 RSC.
- H. Nishide and K. Oyaizu, Science, 2008, 319, 737–738 CrossRef CAS PubMed.
- K. Koshika, N. Sano, K. Oyazu and H. Nishide, Chem. Commun., 2009, 836–838 RSC.
- E. Lebegue, T. Brousse, J. Gabicher, R. Retoux and C. Cougnon, J. Mater. Chem. A, 2014, 2, 8599–8602 RSC.
- X. Wei, W. Xu, M. Vijayakumar, L. Cosimbescu, T. Liu, V. Sprenkle and W. Wang, Adv. Mater., 2014, 26, 7649–7653 CrossRef CAS.
- T. Suga, H. Ohshiro, S. Sugita, K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 1627–1630 CrossRef CAS.
- G. I. Likhtenshtein, J. Yamauchi, S. Nakatsuji, A. I. Smirnov and R. Tamura, Nitroxides Applications in Chemistry, Biomedicine, and Materials Science, Wiley-VCH, Weinheim, 2008 Search PubMed.
- X. Yi, S. Lei, W. Liu, F. Che, C. Yu, X. Liu, Z. Wang, X. Zhou and Y. Zhang, Org. Lett., 2020, 22/12, 4583–4587 CrossRef.
- R. A. Sheldon, I. W. C. E. Arends, G.-J. T. Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774 CrossRef CAS.
- R. Ciriminna, G. Palmisano and M. Pagliaro, ChemCatChem, 2015, 7, 552–558 CrossRef CAS.
- A. Kipke, K. U. Schöning, M. Yusubov and A. Kirschning, Eur. J. Org. Chem., 2017, 6906–6913 CrossRef CAS.
- F. Geneste, C. Moinet, S. Ababou-Girard and F. Solal, New J. Chem., 2005, 29, 1520–1526 RSC.
- A. Michaud, G. Gingras, M. Morin, F. Bland, R. Ciriminna, D. Avnir and M. Pagliaro, Org. Process Res. Dev., 2007, 11, 766–768 CrossRef CAS.
- R. Ciriminna and M. Pagliar, Org. Process Res. Dev., 2010, 14, 245–251 CrossRef CAS.
- A. Hirohata, K. Yamada, Y. Nakatani, I.-L. Prejbeanu, B. Diény, P. Pirro and B. Hillebrands, J. Magn. Magn. Mater., 2020, 509, 166711 CrossRef CAS.
- M. M. Chehimi, Aryl Diazonium Salts, New Coupling Agents in Polymer and Surface Science, Wiley-VCH Verlag & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- W. Choi, S. Endo, K. Oyaizu and H. Nishide, J. Mater. Chem. A, 2013, 1, 2999–3003 RSC.
- D. Q. Fan, J. P. He, W. Tang, J. T. Xu and Y. L. Yang, Eur. Polym. J., 2007, 43, 26–34 CrossRef CAS.
- M. Gilhespy, M. Lok and X. Baucherel, Catal. Today, 2006, 117, 114–119 CrossRef CAS.
- C. Peureux and V. Jouikov, Chem. – Eur. J., 2014, 20, 9357–9366 CrossRef PubMed.
- Z. Wilamowski, M. Solnica, E. Michaluk, M. Havlicek and W. Jantsch, Semicond. Sci. Technol., 2011, 26/6, 064009 CrossRef.
- P. Krukowski, W. Kozlowski, W. Olejniczak, Z. Klusek, M. Puchalski, P. Dabrowski, P. J. Kowalczyk, K. Gwozdzinski and G. Grabowski, Appl. Surf. Sci., 2008, 255, 1921–1928 CrossRef CAS.
- P.-Y. Blanchard, O. Alévêque, T. Breton and E. Levillain, Langmuir, 2012, 28, 1371–1374 Search PubMed.
- V. Jouikov and J. Simonet, J. Appl. Electrochem., 2012, 42/7, 527–537 CrossRef.
- V. Jouikov and J. Simonet, Langmuir, 2012, 28/1, 931–938 CrossRef PubMed.
- J. Simonet and V. Jouikov, Electrochem. Commun., 2014, 38, 65–67 CrossRef CAS.
- O. Swiech, R. Bilewicz and E. Megie, RSC Adv., 2013, 3, 5979–5986 RSC.
- A. C. Herath and J. Y. Becker, Electrochim. Acta, 2008, 53, 4324–4330 CrossRef CAS.
- M. Rafiee, B. Karimi and S. Alizadeh, ChemElectroChem, 2014, 1, 455–462 CrossRef.
- R. A. Green, J. T. Hill-Cousins, R. C. D. Brown, D. Pletcher and S. G. Leach, Electrochim. Acta, 2013, 113, 550–556 CrossRef CAS.
Footnotes |
| † This paper is dedicated to Prof. Jacques Simonet for his significant contribution in the fields of organic electrochemistry and surface functionalization. |
| ‡ Electronic supplementary information (ESI) available: Synthetic details, EIS at TMP and TMP·HCl interfaces, EDS data for the TEMPO-grafted gold interface, and oscillograms of redox switching TEMPO+/TEMPO grafted onto graphite. See DOI: https://doi.org/10.1039/d2cc01520j |
| This journal is © The Royal Society of Chemistry 2022 |