
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An activity-based probe targeting the streptococcal virulence factor C5a peptidase†
Sankarganesh
Krishnamoorthy‡
a,
Andrea K.
Steiger‡
a,
William C.
Nelson
a,
Robert G.
Egbert
a and
Aaron T.
Wright
 *ab
*ab
aBiological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99352, USA. E-mail: Aaron.wright@pnnl.gov
bThe Gene and Linda Voiland School of Chemical Engineering and Bioengineering, Washington State University, Pullman, Washington 99163, USA
First published on 30th June 2022
Abstract
Development of profiling strategies to provide high resolution understanding of enzymes involved in bacterial infections remains an important need. These strategies help resolve enzyme mechanisms of actions and can guide therapeutic development. We have developed a selective new activity-based probe (ABP) targeting a highly conserved surface bound enzyme, C5a peptidase, present in several pathogenic Streptococci. We demonstrate our probe inhibits C5a peptidase activity and enables detection of C5a peptidase expressing pathogens in microbial mixtures. Our profiling strategy selectively labels the pathogen by phenotype and enables specific isolation of the live bacteria providing a route for further in-depth investigation. This study paves the way towards a rapid detection, isolation, and characterization pipeline for existing and emerging strains of most common pathogenic Streptococci.
Several enzymes play critical roles as virulence factors in enabling bacterial pathogens to successfully colonize host cells.1 An understanding of the biochemical functions and the environmental conditions that yield expression of virulence factors will enable improved mechanistic understanding of infectivity and mitigation strategies.2 Enhanced understanding also provides an impetus for development of modern tools for detection, isolation, and characterization of related pathogens and disease prevention and remediation strategies.
The pathogenic members of Group A, B, C and G streptococci3–5 are responsible for several ailments in humans and animals.6 For example, S. pyogenes (Group A streptococcus) alone causes pharyngitis, necrotizing fasciitis, and toxic shock syndrome and is associated with an estimated half million deaths annually. Recently, two new strains of S. pyogenes (type emm43.4) with increased level of resistance to ampicillin, cefotaxime and amoxicillin were discovered.7 Group B strep (GBS) are a common cause of severe infection in newborns,8 and S. dysgalactiae, S. agalactiae and S. suis cause both human and animal infections. Almost all known pathogenic Streptococci, except for S. pneumoniae, express a highly virulent enzyme, C5a peptidase. C5a peptidase is a highly conserved surface bound serine protease,9 which plays a key role in human immune system evasion by cleaving and thus inactivating the human complement (C3) and complement-derived chemotactic factors (C5a and C3b).10,11 It also acts as an adhesion factor during invasion of the host by binding integrin, fibronectin, and epithelial cells.3 Owing to the important roles C5a peptidase plays in pathogenicity, particularly its survival in the host, it has been targeted for vaccine development without success despite decades of research.12 Furthermore, there have been no reports of small molecule inhibitors for C5a peptidase, which may provide a route toward future antibiotic alternatives.
Activity-based probes (ABPs) are uniquely suited for pathogen detection through selective labeling of virulence enzymes in intact cells. ABPs are designed to selectively capture target enzymes while enabling visualization via a reporter moiety.13 When the target enzyme is cytosolic or surface-bound, this imparts distinct optical properties to the associated cell as well. Upon activation, ABPs covalently bind to the target, enabling direct analysis of the enzyme and intact cells by analyses such as fluorescence microscopy, flow cytometry and fluorescent activated cell sorting (FACS) in whole cells and SDS-page and LC-MS based proteomics after lysis. FACS coupled with ABPs is an excellent tool for identification and enrichment of the organism expressing the active target enzyme on a single cell level.14 Previously, ABPs were coupled with FACS for detection and isolation of sub-populations of fixed microbial cells from gut microbiome samples.15 Thus, by choosing a unique enzyme activity specific to pathogenic bacteria, the approach can be extended for rapid labeling, detection, and isolation of the living pathogen associated with the virulence factor.
Identification of pathogens is a preliminary step in a series of steps to determine disease-causing sources before imposing preventive measures.16 Although typical microbiological techniques allow identification and isolation of bacteria, it is tedious and challenging for various reasons:17 high abundance of microbes in nourishing environments, co-occurrence of target pathogen in complex microbial communities, complexity of native medium (e.g. bodily fluids), low abundance of target pathogen, lack of viability, and lack of expression of pathogenic traits during cultivation in the common broths and medias.18 Furthermore, the expression of virulence-associated enzymes generally occurs alongside the expression of other virulence-associated traits to successfully adapt and survive in the immediate environment (e.g. in host).19 Thus, targeting virulence associated enzymes has a broad scope for detection and isolation of bacteria and to study other changes associated with the expression of targeted virulence enzymes. Therefore, rapid methods and technologies for detection and isolation of live pathogens are highly desired to identify pathogen sources, enable faster clinical diagnosis, and for further investigation of new strains and their pathogenicity.20
To design an ABP selective for C5a peptidase, we proposed synthesis of a histidine analog of phenyl phosphonate ester because C5a peptidase is known to cleave the human C5a, a 74 amino acid peptide, at the C-terminal of His-67.9 The histidine side chain is anticipated to impart high selectivity because there are few serine proteases with primary specificity for histidine,21 the phenyl phosphonate ester group is known to react with serine proteases to form stable covalently bound adducts after liberation of phenoxide,22 and the amino group can be coupled with desired reporter moieties such as fluorophores or a biorthogonal reactive group (azide or alkyne).23–25 Though several amino acid analogs of phenyl phosphonate esters have been reported,26 a phosphonate ester analog of histidine has not been synthesized, to the best of our knowledge. Typically, the phenyl phosphonate esters of amino acid analogs have been synthesized from an aldehyde precursor in the presence of benzyl carbamate/acetic acid and subsequent hydrolysis of the carbamate. The commercial inaccessibility of the aldehyde precursor led us to start the synthesis from a nitrile. While organic nitriles have been used to synthesize amino ethyl phosphonic esters using ethyl phosphite with in situ formed aldimines after DIBAL-H reduction,27 synthesis of aryl phosphonate esters from organic nitriles is not known. This is likely because of the poor hydrolytic stability and presence of residual phenols of aryl phosphites synthesis. However, we decided to pursue this route as it offers the ease of a one-pot reaction if successful (Scheme 1). The commercially available nitrile 1 was readily trityl protected (2),28 then reduced in the presence of DIBAL-H in diethyl ether at −20 °C resulting in 5% yield of 3 and recovery of starting material. We assumed this to be a consequence of poor solubility of 2 in diethyl ether. However, our attempts to improve the yield by switching the solvents to THF and dimethoxy ethane failed. 2 readily dissolved in dichloromethane; however, the reduction carried out at −20 and −40 °C resulted in over-reduction of 2 to the amine. Therefore, the reduction and addition of diphenyl phosphite was carried out at −78 °C resulting in an improved yield of 3 (26%). Then, 3 was coupled with 5-hexynoic acid to obtain 4, which was deprotected in the presence of 0.02N HCl in HFIP providing C5a-Al in 88% yield. Finally, C5a-Al was coupled with the fluorophore 5via copper catalyzed click chemistry to get the desired fluorescent probe, C5a-Fl.
 | ||
Scheme 1 Synthesis of C5a-Fl. Reagents and conditions: (a) Et3N, Ph3CCl, DCM, 90%; (b) (1) DIBAL, −78 °C, 2 h; (2) HP(O)(OPh)2, 26% (c) 5-hexynoic acid, HATU, DIEPA, DMF, 24 h, 83% (d) 0.02N HCl in HFIP, 88% (e) CuSO4, sodium ascorbate, H2O/THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4), 17 h, 50%. :4), 17 h, 50%. | ||
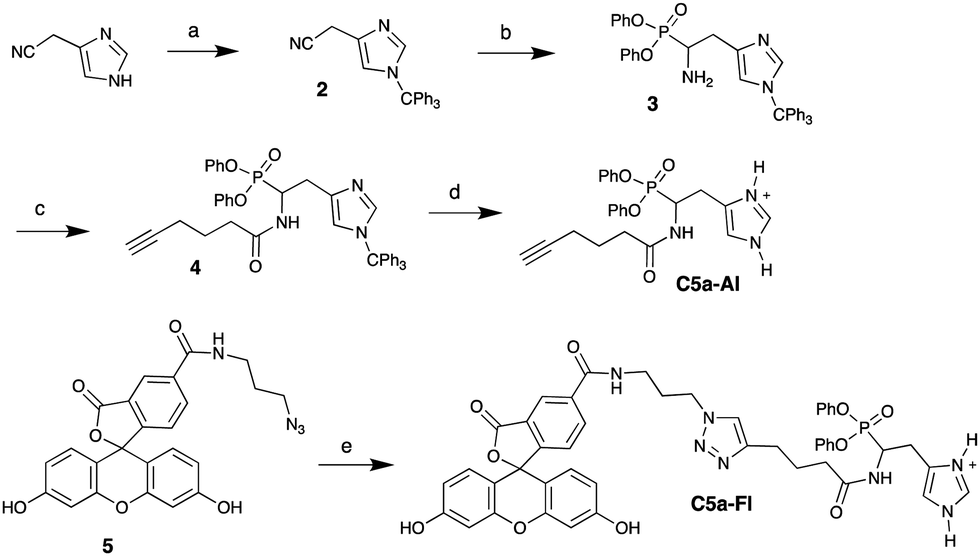
Characterizing C5a peptidase activity has been challenging because of the difficulty in analyzing the native substrate (C5a),29 time-consuming purification from over-expression systems and extraction of membrane bound C5a peptidase from target Gram positive species (see S1, ESI†).30 Thus, a simple and efficient assay to characterize this activity in whole cells is highly desired. Previously, Cleary and Stafslien developed a gel-based assay using a glutathione-S-transferase (GST)-C5a-green fluorescent protein (GFP) fusion as a substrate to study purified C5a peptidase activity.29 Because of the lack of commercially available GST-C5a-GFP, we envisioned a gel-based assay (Fig. 1) using commercially available human C5a tagged with a fusion protein (C5a-Fc; 34.6 kDa) and SyproRuby protein gel stain. Because C5a peptidase is a surface protein, it also provides a route to directly study C5a peptidase activity in whole cells. To test whole-cell detection of C5a peptidase with our probe, we developed an inducible C5a peptidase expression system in E. coli RE100031 following Anderson et al.30 C5a-Fc (>150 ng; 39 kDa, see S2, ESI†) was incubated with the expression system (RE1000/pBAD-scpA), and the proteins within the supernatant were separated by SDS-PAGE and visualized with Sypro Ruby (Fig. 1). A new band, identified in supernatant of RE1000/pBAD-scpA around 30 kDa (also see S3, ESI† for other controls), was analyzed by LC-MS after in-gel trypsin digestion;32 this band was composed of 68% of the peptides from the Fc fragment. The new band formation was also observed with S. pyogenes and S. agalactiae (see S4, ESI†) in similar experiments, confirming C5a peptidase activity in each of these species.
 | ||
| Fig. 1 scpA-E. coli activity on C5a-Fc and SDS-PAGE analysis. | ||
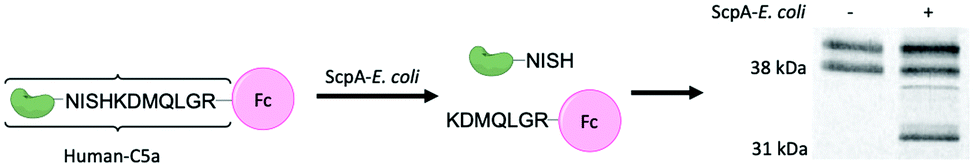
We envisioned the above gel-based assay could be used to show C5a-Fl is targeting C5a peptidase activity. If C5a-Fl inhibits activity, we expect to observe an attenuation of the production of the Fc fragment at 30 kDa. To illustrate inhibition on a gel, first, RE1000 pBAD-scpA was incubated with C5a-Fl at various concentrations, then C5a-Fc was introduced, and the supernatant was analyzed by SDS-PAGE. We observed a decrease in the intensity of the Fc fragment (Fig. 2, see S5, ESI† for related controls) as the probe concentration increased, clearly demonstrating that C5a peptidase activity was inhibited by C5a-Fl. Similar trends were also observed in the presence of C5a-Al (see S6, ESI†).
 | ||
| Fig. 2 (a) Cartoon depicting C5a-Fl binding to C5a peptidase.§ (b) SyproRuby stained SDS-PAGE gel showing change in the intensity of Fc fragment from C5a-Fc cleaved by C5a peptidase activity in the presence of C5a-Fl. | ||
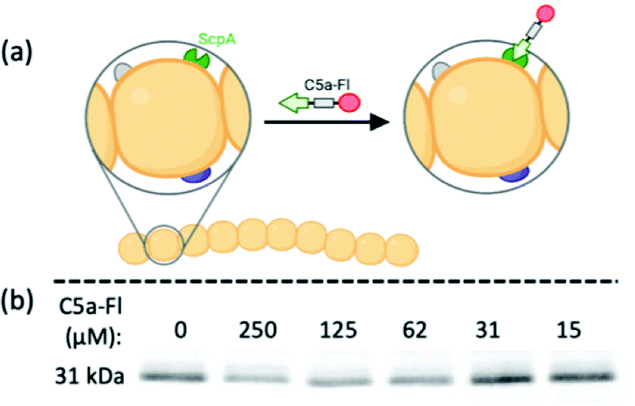
Having established C5a peptidase activity and its inhibition by C5a-Fl in C5a peptidase expressing E. coli, we experimented with probe labeling in RE1000/pBAD-scpA with varied C5a peptidase expression. For this, we took advantage of the tunable PBAD arabinose-inducible promoter system, where an optimum level of expression is achieved when cultured in the presence of arabinose (induced) and the expression is inhibited in the presence of glucose, though a marginal background level of enzyme is still expressed.33 RE1000/pBAD-scpA and relevant controls cultures were incubated with C5a-Fl (125 μM) and characterized by flow cytometry (Fig. 3; see S7, ESI† for histograms). The increase in fluorescence was determined for each sample by normalizing against the fluorescence of the DMSO-only control cultures. As expected, the RE1000 cells lacking pBAD-scpA expression showed limited labeling (minimal increase in FITC fluorescence), and only the RE1000/pBAD-scpA cultures induced with arabinose showed significant (p < 0.0001) labeling by C5a-Fl. The observed labeling agrees with anticipated C5a-Fl selectivity for C5a peptidase.
 | ||
| Fig. 3 Fluorescence increase resulting from selective labeling of RE1000/pBAD-scpA induced by arabinose in the presence of C5a-Fl (125 μM). **** denotes p < 0.0001. | ||
Confident that C5a-Fl selectively labels the ScpA-overexpressing model of C5a peptidase active microbes, we next examined if the probe would label wild type microbes with C5a peptidase activity. To test this, S. pyogenes and S. agalactiae were incubated with C5a-Fl at various concentrations and compared to labeling of S. pneumoniae and E. coli Nissle 1917 as negative controls not containing C5a peptidase. The samples were analyzed by flow cytometry and the fluorescence increase of the samples was determined by normalizing against the fluorescence of the DMSO-only control cultures (Fig. 4; see S8, ESI† for histograms). A statistically significant increase in fluorescence was observed for both S. pyogenes and S. agalactiae at 125 μM compared to both E. coli and S. pneumoniae confirming that C5a-Fl is selective for C5a peptidase-producing wild-type microbes. The amount of probe used may be further reduced when using enantiomerically pure probe mimicking the L-Histidine. Subsequently, viability of the target bacteria were tested after probe incubation and found no significant change from the no probe controls (see S9, ESI†), confirming the possibility of isolation of live bacteria after C5a-Fl labeling.
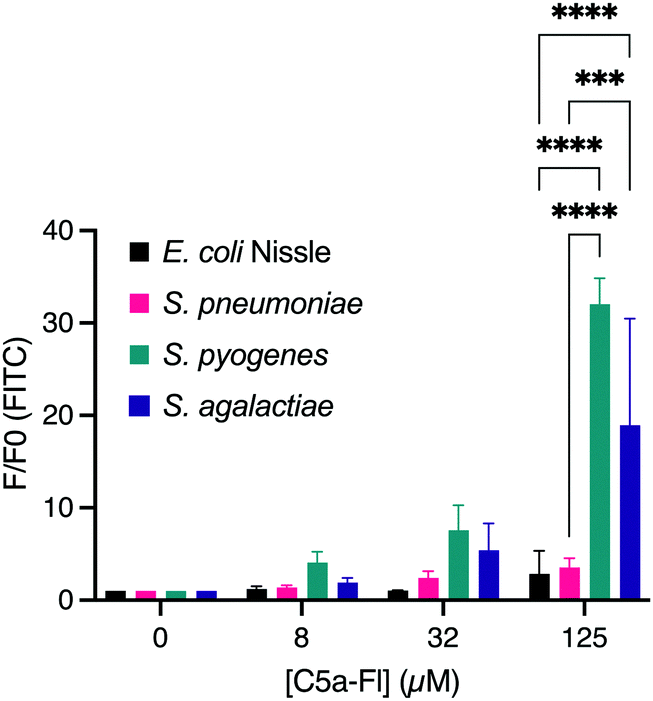 | ||
| Fig. 4 Dose dependent labeling of E. coli Nissle, S. pneumoniae, S. pyogenes and S. agalactiae. *** denotes p <0.001 and **** denotes p < 0.0001. | ||
We next sought to evaluate whether that selectivity would persist when target and nontarget organisms were labeled within the same sample. To perform this, we mixed a suspension of RFP-E. coli (mKate2, details in ESI†) with target microbes (2:1 ratio; Fig. 5a and labeled with C5a-Fl. Analysis of the mixture by both FITC and RFP fluorescence enables discrimination of which species is labeled by C5a-Fl by flow cytometry. Gates were drawn based on unlabeled controls (see S10, ESI†). RFP fluorescence was used to discriminate the two species and FITC fluorescence was used to differentiate probe labeled from unlabeled cells. Following incubation with C5a-Fl, we observed a significant increase in FITC fluorescence for each of the Streptococcus populations, while the E. coli remained unlabeled (Fig. 5a–c). This confirmed the probe's selectivity for Streptoccoci species even within the presence of another microbe, and that activation of the probe by C5a peptidase does not result in off-target labeling.
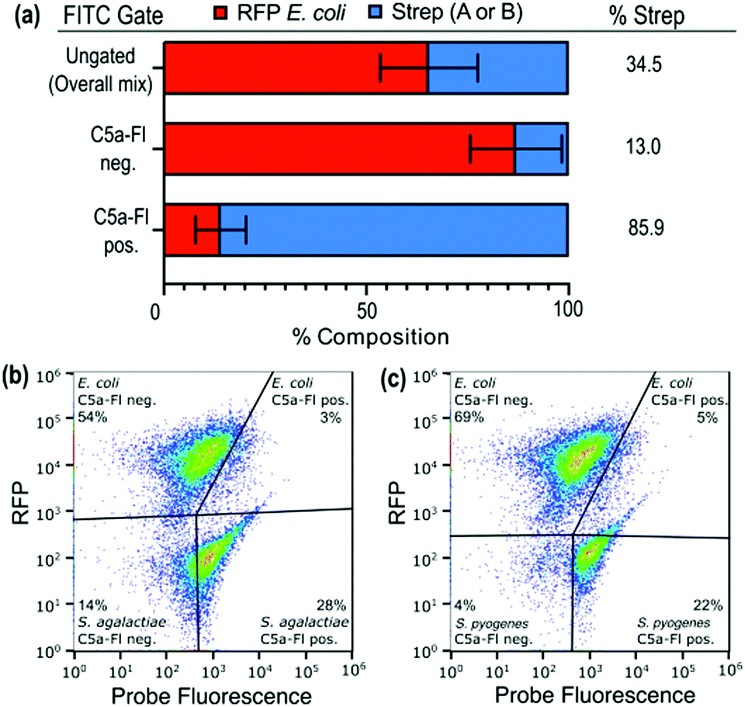 | ||
| Fig. 5 Comparison of the composition of Streptococci and RFP E. coli in a mixture as well as the C5a-Fl positive and negative gates. The C5a-Fl positive gate shows an enrichment of Streptococci species. (a). Flow cytometry data for mixtures with RFP E. coli shown for (b) S. agalactiae and (c) S. pyogenes. Gates were set based on unstained and single-species controls. | ||
In summary, we have developed a selective activity-based probe targeting a virulence enzyme (C5a peptidase) of common pathogenic Streptococci. species, including S. pyogenes and S. agalactiae. Employing C5a-Fl, we have shown inhibition of C5a peptidase activity in whole cells using a simple SDS-PAGE assay, methods for analysis of C5a peptidase activity in a recombinant E. coli system, and selective profiling of viable pathogenic Streptococci in the presence of non-pathogenic bacteria. These results can be further adapted to readily characterize C5a peptidase expression and its role under various environmental conditions. Furthermore, the probe is the first known small molecule inhibitor for C5a peptidase, whose structural components can be adapted for further investigation of clinically relevant small molecule inhibitors. We are currently investigating the labeling capability of our probe in complex environmental and biological samples for selective detection of C5a peptidase expressing Streptococci.
This research was funded by the Defense Advanced Projects Agency (DARPA), grant number HR001118S0025-FoF-FP-001. PNNL is operated by Battelle for the DOE under contract DE-AC06-76RL01830.
Conflicts of interest
The authors declare no conflicts.Notes and references
- V. Pancholi and G. S. Chhatwal, Int. J. Med. Microbiol., 2003, 293, 391–401 CrossRef CAS PubMed.
- A. J. Barr, Essays Biochem., 2018, 62, 619–642 CrossRef PubMed.
- C. Beckmann, J. D. Waggoner, T. O. Harris, G. S. Tamura and C. E. Rubens, Infect. Immun., 2002, 70, 2869–2876 CrossRef CAS PubMed.
- S. A. Lother, W. Demczuk, I. Martin, M. Mulvey, B. Dufault, P. Lagace-Wiens and Y. Keynan, Emerging Infect. Dis., 2017, 23, 1079–1088 CrossRef PubMed.
- P. P. Cleary, J. Peterson, C. Chen and C. Nelson, Infect. Immun., 1991, 59, 2305–2310 CrossRef CAS PubMed.
- W. Krzysciak, K. K. Pluskwa, A. Jurczak and D. Koscielniak, Eur. J. Clin. Microbiol. Infect. Dis., 2013, 32, 1361–1376 CrossRef CAS PubMed.
- K. S. Vannice, J. Ricaldi, S. Nanduri, F. C. Fang, J. B. Lynch, C. Bryson-Cahn, T. Wright, J. Duchin, M. Kay, S. Chochua, C. A. Van Beneden and B. Beall, Clin. Infect. Dis., 2020, 71, 201–204 CrossRef CAS PubMed.
- M. Rosa-Fraile and B. Spellerberg, J. Clin. Microbiol., 2017, 55, 2590–2598 CrossRef CAS PubMed.
- P. P. Cleary, U. Prahbu, J. B. Dale, D. E. Wexler and J. Handley, Infect. Immun., 1992, 60, 5219–5223 CrossRef CAS PubMed.
- N. N. Lynskey, M. Reglinski, D. Calay, M. K. Siggins, J. C. Mason, M. Botto and S. Sriskandan, PLoS Pathog., 2017, 13, e1006493 CrossRef PubMed.
- D. E. Wexler, D. E. Chenoweth and P. P. Cleary, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 8144–8148 CrossRef CAS PubMed.
- J. B. Dale and M. J. Walker, Curr. Opin. Infect. Dis., 2020, 33, 244–250 CrossRef CAS PubMed.
- H. Fang, B. Peng, S. Y. Ong, Q. Wu, L. Li and S. Q. Yao, Chem. Sci., 2021, 12, 8288–8310 RSC.
- L. Chen, L. J. Keller, E. Cordasco, M. Bogyo and C. S. Lentz, Angew. Chem., Int. Ed., 2019, 58, 5643–5647 CrossRef CAS PubMed.
- C. Whidbey, N. C. Sadler, R. N. Nair, R. F. Volk, A. J. DeLeon, L. M. Bramer, S. J. Fansler, J. R. Hansen, A. K. Shukla, J. K. Jansson, B. D. Thrall and A. T. Wright, J. Am. Chem. Soc., 2019, 141, 42–47 CrossRef CAS PubMed.
- C. R. L. M. Sehulster, M. J. Arduino, J. Carpenter, R. Donlan, D. Ashford, R. Besser, B. Fields, W. C. M. M. McNeil, S. Wong, D. Juranek and J. Cleveland, Guidelines for Environmental Infection Control in Health-Care Facilities; U.S. Department of Health and Human Services Centers for Disease Control and Prevention (CDC): Atlanta, GA, 2019 Search PubMed.
- S. G. Mulamattathil, C. Bezuidenhout, M. Mbewe and C. N. Ateba, J. Pathog., 2014, 2014, 371208 Search PubMed.
- P. H. Janssen, P. S. Yates, B. E. Grinton, P. M. Taylor and M. Sait, Appl. Environ. Microbiol., 2002, 68, 2391–2396 CrossRef CAS PubMed.
- M. S. Thomas and S. Wigneshweraraj, Virulence, 2014, 5, 832–834 CrossRef PubMed.
- P. Rajapaksha, A. Elbourne, S. Gangadoo, R. Brown, D. Cozzolino and J. Chapman, Analyst, 2019, 144, 396–411 RSC.
- E. D. Cera, IUBMB Life, 2009, 61, 510–515 CrossRef PubMed.
- A. S. Abuelyaman, D. Hudig, S. L. Woodard and J. C. Powers, Bioconjugate Chem., 1994, 5, 400–405 CrossRef CAS PubMed.
- N. C. Sadler and A. T. Wright, Curr. Opin. Chem. Biol., 2015, 24, 139–144 CrossRef CAS PubMed.
- L. E. Sanman and M. Bogyo, Annu. Rev. Biochem., 2014, 83, 249–273 CrossRef CAS PubMed.
- C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS PubMed.
- M. Maslanka and A. Mucha, Pharmaceuticals, 2019, 12, 86 CrossRef CAS PubMed.
- Z. H. Kudzin and M. W. Majchrzak, J. Organomet. Chem., 1989, 376, 245–248 CrossRef CAS.
- T. V. Hughes and M. P. Cava, Tetrahedron Lett., 1998, 39, 9629–9630 CrossRef CAS.
- D. K. Stafslien and P. P. Cleary, J. Bacteriol., 2000, 182, 3254–3258 CrossRef CAS PubMed.
- E. T. Anderson, M. G. Wetherell, L. A. Winter, S. B. Olmsted, P. P. Cleary and Y. V. Matsuka, Eur. J. Biochem., 2002, 269, 4839–4851 CrossRef CAS PubMed.
- R. G. Egbert, H. S. Rishi, B. A. Adler, D. M. McCormick, E. Toro, R. T. Gill and A. P. Arkin, Nucleic Acids Res., 2019, 47, 3244–3256 CrossRef CAS PubMed.
- A. Shevchenko, H. Tomas, J. Havlis, J. V. Olsen and M. Mann, Nat. Protoc., 2006, 1, 2856–2860 CrossRef CAS PubMed.
- L. M. Guzman, D. Belin, M. J. Carson and J. Beckwith, J. Bacteriol., 1995, 177, 4121–4130 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc01517j |
| ‡ Authors contributed equally. |
| § Fig. 2a created with BioRender.com. |
| This journal is © The Royal Society of Chemistry 2022 |