
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBis[cyclic (alkyl)(amino)carbene] isomers: Stable trans-bis(CAAC) versus facile olefin formation for cis-bis(CAAC)†
Braulio M.
Puerta Lombardi‡
a,
Ethan R.
Pezoulas‡
a,
Roope A.
Suvinen
 b,
Alexander
Harrison
a,
Zachary S.
Dubrawski
a,
Benjamin S.
Gelfand
a,
Heikki M.
Tuononen
*b and
Roland
Roesler
*a
b,
Alexander
Harrison
a,
Zachary S.
Dubrawski
a,
Benjamin S.
Gelfand
a,
Heikki M.
Tuononen
*b and
Roland
Roesler
*a
aDepartment of Chemistry, University of Calgary, 2500 University Drive NW, Calgary, AB T2N 1N4, Canada. E-mail: roesler@ucalgary.ca
bDepartment of Chemistry, Nanoscience Centre, University of Jyväskylä, P.O. Box 35, FI-40014 Jyväskylä, Finland. E-mail: heikki.m.tuononen@jyu.fi
First published on 18th May 2022
Abstract
Isomeric bis(aldiminium) salts with a 1,4-cyclohexylene framework were synthesized. The first isolable bis(CAAC) was prepared from the trans-stereoisomer and its ditopic ligand competency was proven by conversion to iridium(I) and rhodium(I) complexes. Upon deprotonation, the cis-isomer yielded an electron rich olefin via a classic, proton-catalyzed pathway. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond formation from the desired cis-bis(CAAC) was shown to be thermodynamically very favorable and to involve a small activation barrier. Compounds that can be described as insertion products of the cis-bis(CAAC) into the E–H bonds of NH3, CH3CN and H2O were also identified.
C bond formation from the desired cis-bis(CAAC) was shown to be thermodynamically very favorable and to involve a small activation barrier. Compounds that can be described as insertion products of the cis-bis(CAAC) into the E–H bonds of NH3, CH3CN and H2O were also identified.
First reported in 2005,1 cyclic (alkyl)(amino)carbenes (CAACs) have been rapidly incorporated into the main-group and organometallic ligand toolkit. Their exceptional σ-donating and π-accepting abilities led to the isolation of a flurry of compounds of fundamental and applied interest.2 Prominent examples include homoleptic late transition metal compounds in low oxidation states,3 main-group and organoradical spin carriers,4 elements in unusual oxidation states,5 and high-performing transition metal catalysts.6 The growing library of accessible CAACs is facilitating steric and electronic profile tuning of their complexes for tailored applications.7–9 Along with the much used five-membered Me2CAAC,1 CyCAACs,1 and AdCAAC,10 other examples include CAACs incorporating imine or phosphine pendant arms,11 six-membered CAAC-6,12 and bicyclic BiCAACs13 (Chart 1). The steady expansion of the CAAC library was facilitated by the straightforward access to the respective aldiminium-salt precursors starting from ubiquitous building blocks, via an elegant protonation-cyclization-hydroiminiumation sequence reported by Bertrand.14
 | ||
| Chart 1 Selected examples of CAACs. | ||
Despite the growing library of CAAC ligands and the tremendous success of bis(NHC)15 (NHC = N-heterocyclic carbene) and bisphosphine analogs16, no bis(CAAC) has been reported to date. We reasoned that this notable absence could be remedied in few synthetic steps, by formally derivatizing CyCAAC,1 which was shown to be a competent ligand. The cyclohexyl scaffold could be used to build two desirable bis(CAAC)s: A bidentate cis-stereoisomer and a ditopic trans-stereoisomer (Chart 1). Our investigations targeting these derivatives will be reported herein.
Aldiminium precursors 3a and 4a were obtained in gram quantities via standard CAAC-building protocols,14 adapted to accommodate the second CAAC moiety (Scheme 1). A commercially available isomeric mixture of cyclohexane-1,4-dimethanols was converted to cyclohexyl-1,4-dicarboxaldehydes and, following condensation with DippNH2 (Dipp = 2,6-diisopropylphenyl), pure trans-diimine 1a could be isolated in 29% overall yield. Double deprotonation of this precursor with n-butyllithium followed by reaction of the resulting aza-allyl anion with 3-bromo-2-methylpropene generated cis- and trans-2a, which were isolated as a mixture.
 | ||
| Scheme 1 Succession of reactions leading to the synthesis of bis(CAAC) precursors 3a and 4a,b. | ||
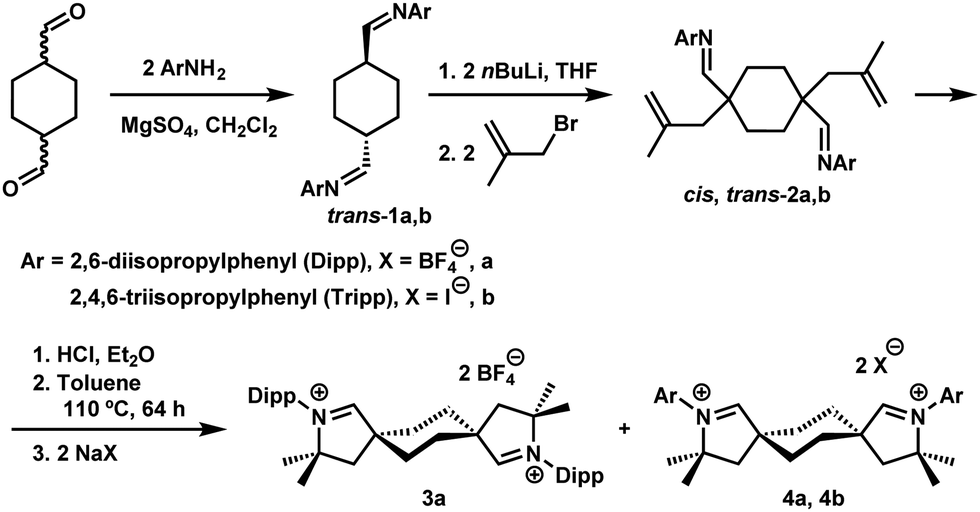
Layering benzene solutions of this mixture with acetonitrile resulted in selective crystallization of trans-2a as large colorless blocks in 10% yield. This compound was then subjected to the hydroiminiumation procedure, leading to dialdiminium tetrafluoroborate salt 3a. cis-Dialdiminium tetrafluoroborate 4a was more conveniently obtained by carrying on the hydroiminiumation reaction with a mixture of cis- and trans-2a. Extraction of the product mixture with CHCl3 and recrystallization by layering CH2Cl2 solutions with hexanes yielded 4a in 67% yield. The configuration of the aldiminium fragments was readily assessed by 1H NMR, based on coupling patterns for the cyclohexylene linker protons (in CD2Cl2, 3a exhibits two doublet resonances at 1.93 and 2.65 ppm and 4a features a pair of multiplets at 2.20 and 2.52 ppm), and confirmed by single-crystal X-ray diffraction (Fig. 1).
 | ||
| Fig. 1 Solid-state structures of the dications in 3a (top) and 4a (bottom) with 50% probability ellipsoids and hydrogen atoms omitted for clarity. Selected bond lengths [Å] and angles [°]: 3a: C1–N1 1.272(4), C1–C2 1.491(4); N1–C1–C2 114.7(2); 4a: C1–N1 1.275(2), C1–C2 1.482(3); N1–C1–C2 114.76(17). | ||
Addition of two equivalents of potassium hexamethyldisilazide (KHMDS) to trans-aldiminium salt 3a in THF produced the expected dicarbene 5 (Scheme 2 and Fig. 2), displaying a characteristic 13C NMR resonance corresponding to the carbenic carbons at 315.2 ppm in C6D6. Under an inert atmosphere, 5 could be handled at room-temperature and no decomposition was observed by 1H NMR after storing the solid at −40 °C for a month. The ditopic nature of 5 was probed via reaction with IrCl(CO)(PPh3)2 or [Rh(cod)Cl]2 in benzene, which yielded complexes 6 (Fig. 3) and 7 (Fig. S67, ESI†), respectively, as yellow, crystalline precipitates. The four ligands in 6 adopt a square planar coordination geometry at iridium, with PPh3trans to the carbene, as previously observed in analogous IrCl(CO)(NHC)(PPh3) complexes.17 The spectral signature of 6 (δcarbene 255 ppm, δCO 174 ppm, δP 22 ppm, νCO 1950 cm−1) is also similar to that of IrCl(CO)(NHC)(PPh3) (δcarbene 178 ppm, δCO 171 ppm, δP 25 ppm, νCO 1945 cm−1).17
 | ||
| Scheme 2 Synthesis of free dicarbene 5 and its metal complexes 6 and 7. | ||
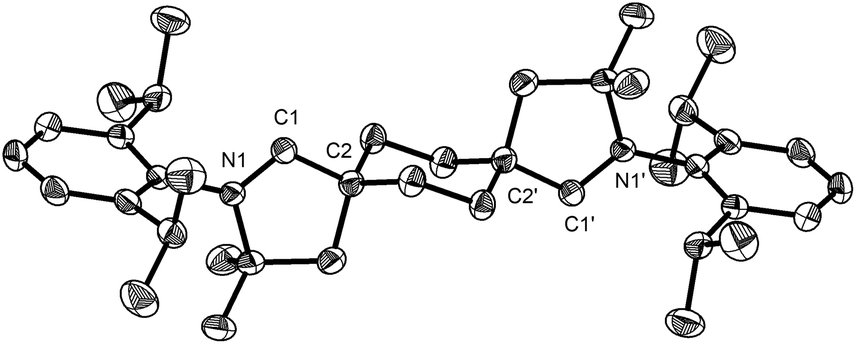 | ||
| Fig. 2 Solid-state structure of 5 with 50% probability thermal ellipsoids and hydrogen atoms omitted for clarity. Selected bond lengths [Å] and angles [°]: N1–C1 1.3065(16), C1–C2 1.5232(17); N1–C1–C2 105.92(10). | ||
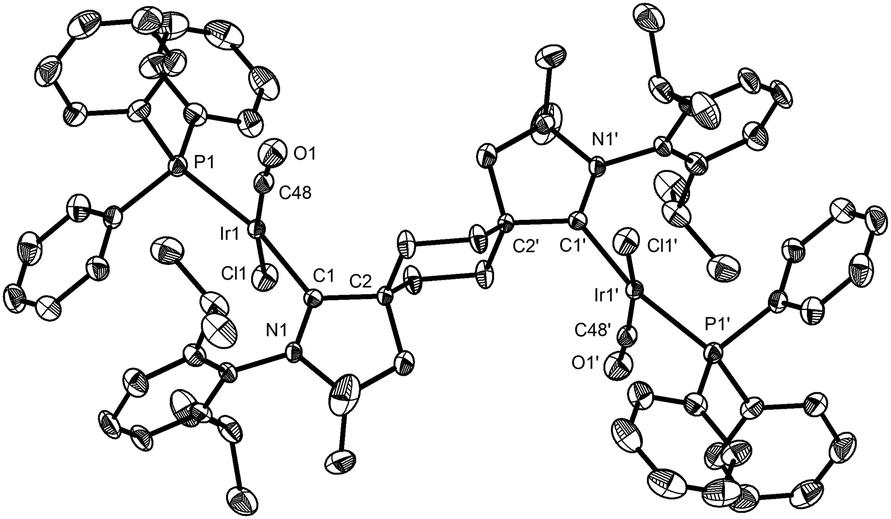 | ||
| Fig. 3 Solid-state structure of 6 with 50% probability thermal ellipsoids and hydrogen atoms omitted for clarity. Selected bond lengths [Å] and angles [°]: Ir1–C1 2.035(3), Ir1–P1 2.3111(8), O1–C48 1.100(3), C1–Ir1–P1 168.68(7), C48–Ir1–Cl1 168.69(9), N1–C1–C2 109.3(2). | ||
Addition of two equivalents of Et3N, iPr2NEt, LiHMDS, LDA, MeLi or Me3SiCH2Li to a suspension 4a in THF of led to intractable mixtures. When lithium-2,2,6,6-tetramethylpiperidide (LiTMP) was employed, an elimination reaction took place, regenerating cis-2a. A similar behavior was reported by Bertrand for CAAC-6.12 Immediately upon addition of two equivalents of KHMDS to 4a at −78 °C, (Fig. S55 and S56, ESI†), a singlet resonance was detected at 318.0 ppm by 13C NMR, suggesting the formation of a CAAC. It rapidly disappeared upon warming to −38 °C with concurrent emergence of a new set of resonances suggestive of a less symmetric compound. In the 1H NMR spectrum, a singlet at 5.08 ppm, within the range observed for protonated NHC-CAAC dimers (4.90–5.32 ppm in CD3CN;18 4.58, 4.72 ppm in CDCl3),19 was assigned to the protonated olefin intermediate [(8)H]BF4 (Scheme 3). Ultimately, warming up the mixture to room temperature led to the formation of a higher-symmetry compound, confirmed via X-ray crystallography to be olefin 8 (Fig. S68, ESI†); whole-molecule disorder precluded a detailed discussion of bonding parameters. Similar electron-rich olefins were recently reported by Sarkar.20 Solutions of 8 in toluene were stable up to 110 °C and the solid could be handled in air. The substantial steric crowding in 8 forces the nitrogen atoms to become pyramidalized (sum of nitrogen bond angles 351.4(4)°). Furthermore, two methyl groups within the Dipp fragments are forced into close proximity to the opposing aryl ring, giving rise to a strongly shielded 1H NMR resonance at −0.02 ppm.
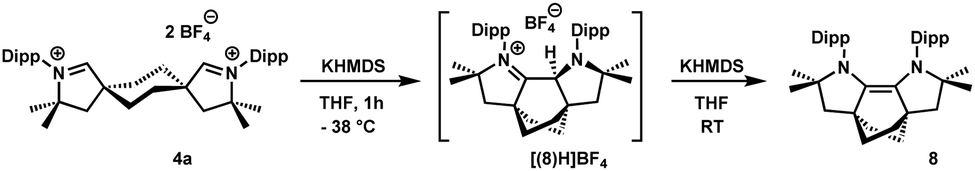 | ||
| Scheme 3 Synthesis of 8via [(8)H]BF4. | ||
DFT studies showed that, upon single deprotonation of 4a to a free mono-CAAC, the cyclohexane backbone readily changes conformation from chair to twist boat ( , Fig. S72, ESI†). A second conformational change from twist boat to boat, accompanied by the formation of a C–C bond to give [8(H)]+, is similarly facile (
, Fig. S72, ESI†). A second conformational change from twist boat to boat, accompanied by the formation of a C–C bond to give [8(H)]+, is similarly facile (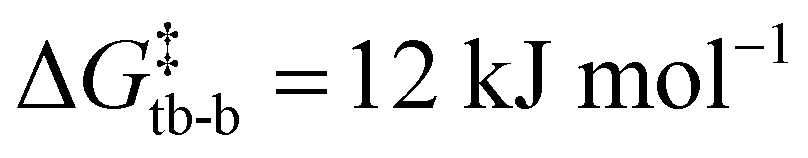 ). The two-step transformation is overall exergonic (ΔG = −72 kJ mol−1) and expected to take place rapidly even at −38 °C due to the associated small energy barriers. The calculated 1H NMR chemical shifts of [8(H)]+ are in good agreement with the experimental values, with a characteristic singlet at 5.91 ppm vs. 5.08 ppm observed experimentally for the protonated olefin. A hypothetical cis-bis(CAAC) resulting from double deprotonation of 4a gave a potential energy surface similar to single deprotonation, with greater energy barriers for conformational changes of the cylochexane ring (
). The two-step transformation is overall exergonic (ΔG = −72 kJ mol−1) and expected to take place rapidly even at −38 °C due to the associated small energy barriers. The calculated 1H NMR chemical shifts of [8(H)]+ are in good agreement with the experimental values, with a characteristic singlet at 5.91 ppm vs. 5.08 ppm observed experimentally for the protonated olefin. A hypothetical cis-bis(CAAC) resulting from double deprotonation of 4a gave a potential energy surface similar to single deprotonation, with greater energy barriers for conformational changes of the cylochexane ring ( and
and  , Fig. S73, ESI†). This likely stems from the increased repulsion associated with two carbon atoms with lone pairs. The formation of 8 from the cis-bis(CAAC) is overall highly exergonic (ΔG = −262 kJ mol−1) as a CC double bond is formed between the two carbenic carbon atoms.
, Fig. S73, ESI†). This likely stems from the increased repulsion associated with two carbon atoms with lone pairs. The formation of 8 from the cis-bis(CAAC) is overall highly exergonic (ΔG = −262 kJ mol−1) as a CC double bond is formed between the two carbenic carbon atoms.
The calculations support the intermediacy of [8(H)]BF4 en route from 4a to 8. Furthermore, even though double deprotonation of 4a could take place prior to conformational changes, the energy barriers associated with the cyclohexane ring flip are minor. This suggests that the cis-bis(CAAC) species obtained via double deprotonation of 4a is not isolable under any conditions. While these results might initially seem surprising, they become less so upon comparison with data calculated for the dimerization of Me2CAAC, which show ΔGdimer = −75 and −99 kJ mol−1 for cis and trans product geometries, respectively. Thus, dimerization of two CAACs is always thermodynamically favored but generally kinetically blocked by very high activation barriers (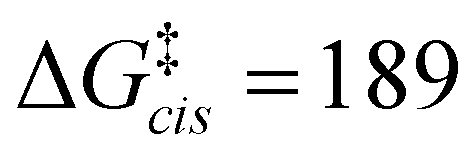 and
and 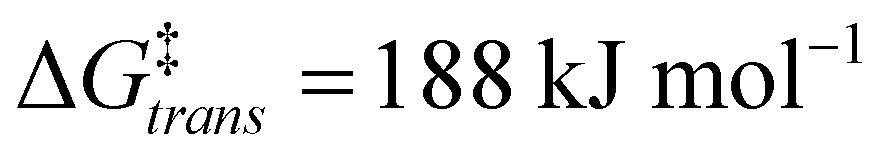 ) that arise in large part from the entropic penalty of dimerization (−TΔS = 81 kJ mol−1). No such penalty exists when the two CAAC moieties are part of the same molecule, which supports the facile formation of 8.
) that arise in large part from the entropic penalty of dimerization (−TΔS = 81 kJ mol−1). No such penalty exists when the two CAAC moieties are part of the same molecule, which supports the facile formation of 8.
Cyclic voltammetry of 8 in THF revealed two reversible redox waves (E1/2 = −0.32 and 0.49 vs. Fc/Fc+, Fig. S59, ESI†), corresponding to the oxidation of 8 to its radical cation and further to the dication, respectively. Both oxidations are anodically-shifted in comparison to the values reported by Sarkar,20 potentially due to the inductive effect of the N-aryl substituent in 8 leading to a less electron rich system. The C2-symmetric radical cation was isolated as purple tetrafluoroborate salt 9 following the oxidation of 8 with [Ph3C][BF4]. It was characterized by EPR (Fig. S60, ESI†) and its structure was confirmed by X-ray crystallography (Fig. 4).
 | ||
| Fig. 4 Solid-state structure of the cation in 9 with 50% thermal ellipsoids and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): C1–C7 1.409(3), C1–N1 1.357(3), C7–N2 1.360(3). | ||
Attempting to destabilize the electron rich olefin 8 in favor of a free cis-bis(CAAC), the Dipp substituents in 4a were replaced with Tripp (2,4,6-triisopropylphenyl) (Scheme 1). However, deprotonation of 4b also led to CC bond formation and this chemistry will not be detailed here. We then turned our attention to complex formation directly from 4a, by employing metal reagents featuring Brønsted-basic ligands (Scheme 4). The reaction of NHC salt precursors with Cu2O or Ag2O to yield the corresponding metal complexes has been extensively explored.21 Extrapolation of this method to CAACs is less common,22 arguably due to the weaker acidity of aldiminium-CAAC salts. Heating an acetonitrile solution of 4a with Cu2O over several days in an NMR tube resulted in crystallization of colorless blocks that were identified as ether 12 by single-crystal X-ray diffraction (Fig. S71, ESI†). The structure is reminiscent of the (Me2CAACH)2O ether obtained as a side-product while preparing (Me2CAAC)2Ge.23 Refluxing 4a and Fe(HMDS)2 in acetonitrile led to crystallization of 11 (Fig. S65, ESI†). The presence of (Me3Si)2NH in the NMR of the product mixture suggests that deprotonation may indeed take place. However, scaling up the reaction led to the isolation of 10 instead (Fig. S70, ESI†). The origin of the nitrogen atom is presumably (Me3Si)2NH, a byproduct from the reaction of Fe(HMDS)2 and 4a. Attempted complex formation using Ca(HMDS)2, Mg(SiTMS3)2, Pd(OAc)2, or [PtMe2(μ-SMe2)]2 gave intractable mixtures.
 | ||
| Scheme 4 Reactivity of 4a towards transition metal complexes with Brønsted-basic ligands. | ||
In conclusion, two isomeric CAAC-aldiiminium salts 3a and 4a, derived from the same parent aldehydes, were synthesized on multi-gram scale. trans-Stereoisomer 3a was doubly deprotonated to yield the first isolable bis(CAAC) 5, which proved to easily form dinuclear metal complexes, as exemplified by iridium complex 6 and rhodium complex 7. Upon double deprotonation with KHMDS, cis-stereoisomer 4a formed the electron-rich olefin 8. Experimental and computational studies suggest the process follows the classic Lewis-acid catalyzed NHC dimerization pathway. DFT calculations showed that the intramolecular CC bond formation in a free cis-bis(CAAC) derived from 4a is highly exergonic (−262 kJ mol−1vs. −75 kJ mol−1 for cis-dimerization of Me2CAAC) and involves, for entropic reasons, a small activation barrier that can be lowered even more by proton catalysis. Reaction of 4a with metal complexes featuring Brønsted-basic ligands led to the identification of 10, 11, and 12, which can be described as insertion products of a bis(CAAC) into the N–H, C–H and O–H bonds of NH3, CH3CN and H2O, respectively. Whether their formation involves a free CAAC intermediate that has been observed at low-temperature by NMR, as described for mono(CAAC)s,24 or the transient formation of metal complexes, remains to be investigated. 17 years after the first report of a CAAC ligand, our study adds the first bis(CAAC) ligand to the organometallic toolkit.
Financial support was provided by the Universities of Calgary and Jyväskylä, as well as the Natural Sciences and Engineering Research Council of Canada (NSERC) in the form of Discovery Grant #2019-07195 to R. R. The project received funding from the European Research Council under the EU's Horizon 2020 programme (grant #772510 to H. M. T.). A. H. and Z. S. D. acknowledge funding from the Canada First Research Excellence Fund (CFREF). Computational resources were provided by the Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533).
Conflicts of interest
There are no conflicts to declare.References
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed.
- M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed; M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef PubMed; V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef PubMed; S. K. Kushvaha, A. Mishra, H. W. Roesky and K. C. Mondal, Chem. – Asian J., 2022, e2021013 Search PubMed.
- S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–369 CrossRef CAS PubMed.
- M. M. Hansmann, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2546–2554 CrossRef CAS PubMed; M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2206–2213 CrossRef PubMed; T. Ullrich, P. Pinter, J. Messelberger, P. Haines, R. Kaur, M. M. Hansmann, D. Munz and D. M. Guldi, Angew. Chem., Int. Ed., 2020, 59, 7906–7914 CrossRef PubMed.
- M. Arrowsmith, H. Braunschweig, M. A. Celik, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, T. Kramer, I. Krummenacher, J. Mies, K. Radacki and J. K. Schuster, Nat. Chem., 2016, 8, 890–894 CrossRef CAS PubMed; W. Lu, Y. Li and R. Kinjo, J. Am. Chem. Soc., 2019, 141, 5164–5168 CrossRef PubMed.
- M. P. Wiesenfeldt, Z. Nairoukh, W. Li and F. Glorius, Science, 2017, 357, 908–912 CrossRef CAS PubMed; J. Morvan, M. Mauduit, G. Bertrand and R. Jazzar, ACS Catal., 2021, 11, 1714–1748 CrossRef.
- Y. Gao, S. Yazdani, A. Kendrick IV, G. P. Junor, T. Kang, D. B. Grotjahn, G. Bertrand, R. Jazzar and K. M. Engle, Angew. Chem., Int. Ed., 2021, 60, 19871–19878 CrossRef CAS PubMed.
- D. Pichon, M. Soleilhavoup, J. Morvan, G. P. Junor, T. Vives, C. Crevisy, V. Lavallo, J. M. Campagne, M. Mauduit, R. Jazzar and G. Bertrand, Chem. Sci., 2019, 10, 7807–7811 RSC.
- J. Morvan, F. Vermersch, Z. Zhang, L. Falivene, T. Vives, V. Dorcet, T. Roisnel, C. Crevisy, L. Cavallo, N. Vanthuyne, G. Bertrand, R. Jazzar and M. Mauduit, J. Am. Chem. Soc., 2020, 142(47), 19895–19901 CrossRef CAS PubMed.
- V. Lavallo, G. D. Frey, S. Kousar, B. Donnadieu and G. Bertrand, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13569–13573 CrossRef CAS PubMed.
- J. Chu, D. Munz, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 7884–7887 CrossRef CAS PubMed.
- C. M. Weinstein, G. P. Junor, D. R. Tolentino, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 9255–9260 CrossRef CAS PubMed.
- E. Tomás-Mendivil, M. M. Hansmann, C. M. Weinstein, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 7753–7756 CrossRef PubMed.
- R. Jazzar, R. D. Dewhurst, J.-B. Bourg, B. Donnadieu, Y. Canac and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 2899–2902 CrossRef CAS PubMed; R. Jazzar, J.-B. Bourg, R. D. Dewhurst, B. Donnadieu and G. Bertrand, J. Org. Chem., 2007, 72, 3492–3499 CrossRef PubMed.
- M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS PubMed; M. Poyatos and E. Peris, Dalton Trans., 2021, 50, 12748–12763 RSC.
- In Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, Wiley, New York, 2012 CrossRef CAS PubMed; A. L. Clevenger, R. M. Stolley, J. Aderibigbe and J. Louie, Chem. Rev., 2020, 120, 6124–6196 CrossRef CAS PubMed.
- C.-F. Fu, Y.-H. Chang, Y.-H. Liu, S.-M. Peng, C. J. Elsevier, J.-T. Chen and S.-T. Liu, Dalton Trans., 2009, 6991–6998 RSC.
- D. Munz, J. Chu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2016, 55, 12886–12890 CrossRef CAS PubMed; J. Messelberger, A. Grünwald, S. J. Goodner, F. Zeilinger, P. Pinter, M. E. Miehlich, F. W. Heinemann, M. M. Hansmann and D. Munz, Chem. Sci., 2020, 11, 4138–4149 RSC.
- D. Mandal, R. Dolai, R. Kumar, S. Suhr, N. Chrysochos, P. Kalita, R. S. Narayanan, G. Rajaraman, C. Schulzke, B. Sarkar, V. Chandrasekhar and A. Jana, J. Org. Chem., 2019, 84, 8899–8909 CrossRef CAS PubMed.
- M. K. Nayak, S. Suhr, N. Chrysochos, H. Rawat, C. Schulzke, V. Chandrasekhar, B. Sarkar and A. Jana, Chem. Commun., 2021, 57, 1210–1213 RSC.
- I. J. B. Lin and C. S. Vasam, Coord. Chem. Rev., 2007, 251, 642–670 CrossRef CAS; M. R. L. Furst and C. S. J. Cazin, Chem. Commun., 2010, 46, 6924 RSC.
- Y. D. Bidal, O. Santoro, M. Melaimi, D. B. Cordes, A. M. Z. Slawin, G. Bertrand and C. S. J. Cazin, Chem. – Eur. J., 2016, 22, 9404–9409 CrossRef CAS PubMed.
- Y. Li, K. C. Mondal, H. W. Roesky, H. Zhu, P. Stollberg, R. Herbst-Irmer, D. Stalke and D. M. Andrada, J. Am. Chem. Soc., 2013, 135, 12422–12428 CrossRef CAS PubMed.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2131007–2131017. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc01476a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |